

AI바라기의 인공지능
protein : 논문 리뷰 : General Multimodal Protein Design Enables DNA-Encoding of Chemistry 본문
protein : 논문 리뷰 : General Multimodal Protein Design Enables DNA-Encoding of Chemistry
AI바라기 2026. 4. 11. 14:55용어 설명
- Co-design: 단백질의 서열(sequence)과 3D 구조(structure)를 순차적이 아닌 완벽하게 동시에 생성하는 방법. (기존 모델들의 한계인 Inverse folding의 단점을 극복하는 핵심 개념)
- Theozyme (Theoretical Enzyme): 양자화학 계산을 통해 미리 정의된 촉매 반응 전이 상태와 이를 안정화하는 핵심 아미노산 잔기들의 고정된 3D 기하학적 배치. (기존 효소 설계의 필수 조건이었으나 DISCO는 이를 탈피함)
- Inverse folding: 단백질의 백본(backbone) 구조를 먼저 생성한 뒤, 그 구조로 접힐 수 있는 아미노산 서열을 나중에 예측해 끼워 넣는 순차적 방식.
- Reactive intermediate: 화학 반응 경로 중간에 일시적으로 생성되는 불안정한 중간 생성물. (DISCO는 전체 전이 상태 대신 오직 이 중간체 구조만을 조건으로 삼아 효소를 설계함)
- Feynman-Kac Corrector (FKC): 생성 모델을 다시 학습(retraining)시키지 않고도, inference 단계에서 특정 목적 함수(reward)를 만족하는 방향으로 샘플링 분포를 직접 조향(steering)하는 수학적 방법론.
- Co-designability: 모델이 생성한 서열을 구조 예측 모델(ESMFold 등)로 다시 접어보았을 때, 모델이 원래 의도했던 생성 3D 구조와 2.0 옹스트롬(Angstrom) RMSD 이내로 일치하는 비율. 모델의 신뢰도를 나타내는 핵심 지표.
- Carbene transferase: 자연계 효소에는 없는 탄소-탄소, 탄소-헤테로원자 결합을 형성하여 새로운 화학 물질을 합성할 수 있는 인공 촉매.
Purpose of the Paper
- 기존 한계 1: Theozyme 의존성. 기존 효소 설계 AI(RFdiffusion 등)는 작동 메커니즘을 완벽히 알아야만 설정 가능한 고정된 아미노산 기하구조(Theozyme)를 미리 제공해야만 작동했음. 이로 인해 메커니즘이 불확실하거나 자연계에 아예 존재하지 않는 완전히 새로운 구조의 활성 부위(active-site)를 발견하는 것이 불가능했음.
- 기존 한계 2: Sequential Pipeline의 한계. 뼈대(structure)를 먼저 만들고 서열(sequence)을 맞추는 Inverse folding 방식은, 백본이 형성되는 가장 중요한 초기 단계에 서열 기반의 화학적 특성 목적 함수를 개입시킬 수 없어 생성물의 물리화학적 현실성이 떨어짐.
- 새로운 접근 방식: DISCO는 사전 정의된 잔기 모티프 없이, 오직 타겟 생체 분자(arbitrary biomolecules) 주변에서 sequence와 structure를 완벽히 동시에 생성하는 Multimodal Co-design framework를 제시하여, 진화가 한 번도 탐색하지 못한 미지의 DNA 인코딩 화학 반응 공간을 개척하고자 함.
Key Contributions
- Unified Multimodal Generative Process 구축: 서열을 위한 masked discrete diffusion과 좌표를 위한 continuous diffusion을 병렬로 결합한 단일 neural network 설계.
- Novelty: Cross-modal recycling 메커니즘을 도입하여, 서열 예측은 점진적으로 나타나는 구조적 특징에 피드백을 받고, 구조 예측은 진화하는 서열 정체성에 적응하는 완벽한 양방향 결합 달성.
- Bypassing Theozymes (사전 정의 없이 De Novo 효소 설계): 전이 상태(transition state) 모델링이나 고정된 촉매 잔기 템플릿 없이, 오직 양자계산(DFT)으로 도출된 Reactive intermediate(반응 중간체)의 기하학적 구조만을 조건으로 주어 효소를 설계.
- Novelty: 인간의 편견(assumptions)이 개입된 잔기 배치 제약을 완전히 벗어나, 모델이 스스로 완전히 새롭고 상호 보완적인 활성 부위 기하구조를 탐색하고 창조해 냄.
- Multimodal Inference Steering (FKC-MM & FKC-SG 도입): 비싼 보상 오라클이나 비효율적인 필터링(best-of-N) 없이, Inference 단계에서 원하는 특성을 직접 주입.
- Novelty: FKC-MM을 통해 Disulfide bond 개수나 Cation-pi interaction처럼 서열과 구조 모두에 의존하는 복합적 속성을 최적화하고, **FKC-SG (Specificity Guidance)**를 통해 구조가 비슷한 오프타겟(decoy)을 밀어내고 온타겟에만 결합하는 고도의 특이성(specificity)을 달성함.
- Inference-time Sequence Self-correction 기법:
- Novelty: Masked diffusion의 약점인 '초기 잘못된 토큰 확정 시 수정 불가' 문제를 해결하기 위해, 추론 과정 중 다시 마스킹하는 기법과 Entropy adaptive sequence temperature를 도입해 과도하게 확신하는 초기 토큰 분포를 억제하여 Co-designability를 극적으로 향상시킴.
Experimental Highlights
- SOTA In silico Performance: Unconditional monomer design에서 기존 모델(RFdiffusion3, BoltzGen, ESM3 등)을 압도하며 약 90%의 Co-designability 달성 (동시에 가장 높은 서열/구조 다양성 유지). STUDIO-179 벤치마크를 통해 극도로 이질적인 179종의 리간드 및 DNA/RNA 복합체에 대해서도 최고 성능 입증.
- 자연계에 없는 촉매(New-to-nature Biocatalysis)의 실험적 증명: 4가지 고난도 Carbene transfer reactions (alkene cyclopropanation, B-H, C(sp3)-H insertions, spirocyclopropanation)을 수행하는 효소 90개를 in vitro에서 스크리닝하여 실제 촉매 활성 확인.
- 기존 진화 효소를 압도하는 수치적 성과:
- 가장 달성하기 어려운 반응인 C(sp3)-H insertion에서 DISCO의 디자인이 **2,360 TTN (Total Turnover Number)**을 달성. (이는 이전에 무려 14라운드의 극단적 directed evolution을 거쳐 만든 최고 성능 효소 P411-CHF의 2,030 TTN을 디자인 단 한 번에 능가한 수치임).
- B-H insertion에서는 98% yield 및 5,170 TTN 달성 (기존 진화 변이체 2,490 TTN의 두 배 이상).
- 완벽한 구조적 참신성(Novelty)과 진화 가능성(Evolvability):
- 생성된 효소(dCT-H11, dCT-F9 등)의 활성 부위는 AlphaFoldDB의 기존 어떤 구조와도 매칭되지 않음 (RMSD 7 옹스트롬 이상 격차).
- dCT-H11 모델에 Error-prone PCR을 단 1회 적용한 결과, 촉매 활성이 크게 오를 뿐만 아니라 입체선택성(enantioselectivity)이 +49% ee에서 -35% ee로 완전히 역전되는 변이체들이 발견됨. 즉, DISCO가 설계한 단백질이 향후 추가 최적화가 매우 용이한 우수한 fitness landscape에 위치해 있음을 증명.
Limitations and Future Work
- Limitation 1: 초기 입체선택성(Stereoselectivity)의 한계. 현재 설계 기준에 입체선택성이 직접적인 조건으로 포함되지 않아, 생성된 효소들의 초기 enantiomeric excess (ee) 수치가 다소 겸손한 수준임(최대 35%).
- Limitation 2: 전자적으로 극도로 까다로운 반응에서의 한계. Spirocyclopropanation과 같이 화학적 난이도가 극상인 반응의 경우, 촉매 활성을 띠는 variant의 비율이 적고 절대적인 촉매 활성도 다소 낮게 나타남.
- Future Work: Generative discovery(모델의 생성)와 Directed evolution(실험적 진화) 사이의 루프를 완전히 닫고(closing the loop), 더 풍부한 물리화학적 제약 조건(biophysical constraints)을 모델에 학습시킴으로써, 향후에는 특정 입체선택성을 스스로 제어하고 자연계의 진화를 능가하는 초고난도 메커니즘을 지닌 효소를 바로 타겟팅하여 생성하는 방향으로 나아가야 함.
Overall Summary
DISCO는 인간이 미리 정의한 Theozyme이나 3D 모티프 없이, 임의의 화학적 반응 중간체 정보만으로 단백질의 서열과 구조를 완벽하게 동시 생성(Co-design)하는 최초의 범용 다중모달 AI 프레임워크입니다. 이 모델은 단순한 타겟 결합을 넘어, 자연계에 존재하지 않는 극한의 화학 반응을 수십 번의 실험적 진화를 거친 기존 최고 성능의 효소보다 더 높은 효율로 수행하는 완전히 새로운 활성 부위를 창조해냈습니다. 이는 DNA 코드로 프로그램할 수 있는 화학적 합성의 범위를 폭발적으로 넓히며, 합성 생물학, 친환경 화학 공정, 차세대 신약 개발 분야에서 인간의 의도대로 즉각 작동하는 맞춤형 생체 촉매를 무한히 설계할 수 있는 새로운 시대를 엽니다.
쉬운 설명
집을 지을 때(효소 설계), 기존 AI 모델들의 방식은 "철골(Structure)을 먼저 다 세워 놓고, 거기에 맞춰서 억지로 벽돌(Sequence)을 끼워 넣는 방식"이었습니다. 게다가 반드시 "부엌은 무조건 이 모양이어야 해(Theozyme)"라는 고정관념이 있어야만 설계가 가능했죠.
하지만 DISCO는 다릅니다. "요리할 재료(화학 반응 중간체)" 하나만 딱 던져주면, 그 재료를 가장 완벽하게 요리할 수 있는 완전히 새로운 형태의 부엌과 집 전체를 동시에(Co-design) 지어냅니다. 설계도를 실시간으로 수정해가며(Self-correction) 뼈대와 벽돌을 함께 쌓아 올리기 때문에, 인간이 상상조차 하지 못했던 독창적이고 엄청나게 효율적인 맞춤형 화학 공장을 단번에 뚝딱 만들어내는 마법의 건축가와 같습니다.
DISCO 모델에게 일을 시킬 때 상황은 이렇습니다.
인간 (Input): "내가 지금 특이하게 생긴 **화학 물질(원자 C, N, O와 이들의 결합 정보)**을 줄게. 이 물질은 시퀀스가 아니라 이렇게 복잡한 3D 입체 화학 구조를 가지고 있어."
AI (DISCO): "알았어. 이 화학 물질의 생김새(원자/결합)를 분석했어. 이 물질을 완벽하게 감싸 안고 화학 반응을 일으킬 수 있는 단백질을 만들어 줄게. 자, 여기 새로 만든 단백질의 **시퀀스(아미노산 배열)**와 3D 뼈대 모양이야 (Output)."
[인풋]
타켓 분자 정보(초기 3d 좌표랑, 결합정보), 길이, 초기 노이즈(완전히 랜덤한 3D 스트럭쳐 좌표, 마스킹된 시퀀스)
[출력]
단백질 서열, 단백질 3D 좌표 구조, 미세 조정된 타겟의 3D 좌표 구조
[동작 방식] (※ 디퓨전 모델이므로, 아래의 전체 과정은 '초기 노이즈 상태'에서 출발해 노이즈가 완전히 깎일 때까지 타임스텝을 거슬러 올라가며 수십~수백 번 반복됨)
타겟 분자정보로 1차원 싱글 리프레젠테이션과 2차원 페어 리프레젠테이션을 만듦. 이때 Pair 표현에는 타겟과 단백질 간의 상대적 위치(Relative position)와 분자의 화학적 결합 구조(Token bonds) 정보가 직접 더해짐.
PLM 사용해서 현재 스텝의 (노이즈 낀) 시퀀스를 읽어 어텐션 가중치를 뽑아서 2차원 페어 텐서에 Add 해버림.
그럼 서열 정보가 더해진 2차원 페어를 알파폴드(Pairformer) 블록에 통과시켜서 기하학적 정보 업데이트.
서열은 다시 서열 임베딩으로 추출하고, 3D 좌표는 구조 임베딩으로 추출.
이 두 임베딩을 1차원 텐서에 Add 해버려서 두 모달리티를 병합함.
페어포머 통과한 3D 구조 정보로 다시 거리 맵을 만들어서 2차원 정보 업데이트.
[노이즈 조건 부여] 현재 스텝에서 시퀀스와 3D 구조에 각각 **'노이즈가 얼마나 남아있는지(Time-step 정보)'**를 임베딩으로 변환해서 1차원 텐서에 더해줌. (이걸 통해 모델이 현재 스텝에서 노이즈를 얼마나 깎아내야 하는지 기준점을 잡음)
시간(노이즈) 정보까지 더해진 후, 원자 단위 정보를 뭉쳐서 아미노산 단위 정보로 압축.
디퓨전 트랜스포머 통과해서 글로벌 컨텍스트 뽑음.
그 텐서는 디코더로 들어가서 두 갈래로 실행됨.
기존 노이즈 낀 3D 좌표에서 원자들이 얼마나 이동해야 할지(업데이트량)를 계산해서 3D 구조를 맞춤.
그리고 또 실행해서 각 자리에 아미노산을 예측해서 시퀀스를 맞춤.
[방향 교정] 이 예측값이 확정되기 직전, 리워드 모델(FKC)을 이용해서 목표에 더 부합하게 텐서의 그래디언트를 살짝 틀면서 원하는 목표(강한 결합 등)로 유도함.
[반복] 이렇게 유도된 방향대로 노이즈를 한 꺼풀 깎아낸 뒤, 그 결과를 다시 다음 스텝의 인풋으로 집어넣어 위 과정을 반복함. 노이즈가 0이 되면 최종 출력 완성.
Abstract
진화는 효소적 다양성을 위한 놀라운 엔진이지만, 진화가 탐구한 화학적 영역은 DNA가 인코딩할 수 있는 범위의 아주 좁은 부분에 불과합니다. Deep generative models는 리간드와 결합하는 새로운 단백질을 design할 수 있지만, 사전에 촉매 잔기를 지정하지 않고 효소를 생성한 model은 아직 없습니다.
우리는 임의의 생체 분자 주변의 단백질 sequence와 3D structure를 co-designs하는 multimodal model인 DISCO와, 두 modalities 전반에 걸쳐 objectives를 optimize하는 inference-time scaling methods를 소개합니다.
반응 중간체에만 Conditioned된 상태에서, DISCO는 새롭고 독창적인 활성 부위 기하학적 구조를 가진 다양한 헴 효소(heme enzymes)를 design합니다. 이러한 효소들은 알켄 사이클로프로판화, 스피로사이클로프로판화, B-H 및 $C(sp^3)-H$ insertions를 포함하여 자연에 존재하지 않는 카르벤 전달 반응을 촉매하며, 이는 기존에 조작된 효소들의 활성을 뛰어넘는 높은 효율을 보여줍니다.
선택된 design에 대한 무작위 돌연변이 유발은 지향성 진화를 통해 효소의 활성이 더욱 향상될 수 있음을 추가로 확인시켜 주었습니다. 진화 가능한 효소로 향하는 scalable 경로를 제공함으로써, DISCO는 유전적으로 인코딩 가능한 변환의 잠재적 범위를 크게 넓혀줍니다.
Introduction
효소는 온화한 조건에서 높은 효율과 특이성으로 화학 반응을 촉매하여, 화학 제조, 약물 합성 및 새로운 치료 양식에서의 응용을 뒷받침합니다. 효소 공학은 전통적으로 원하는 기능을 향해 유익한 sequence 변화를 축적하기 위해 돌연변이 및 스크리닝의 반복적인 과정에 의존해 왔습니다. 이는 효소가 자연에 이전에 알려지지 않은 기능을 수행할 수 있게 하는 진화적 과정입니다. 그러나 이러한 모든 캠페인에는 측정 가능한 활성을 가진 초기 단백질이 필요합니다. 잘 연구된 변환의 경우 천연 단백질 또는 그와 유사한 변이체 중에서 적합한 출발점을 찾을 수 있습니다. 하지만 원해지는 많은 자연계에 존재하지 않는 화학 반응에 대해서 자연은 명확한 후보를 제공하지 않습니다. 이를 식별하는 것은 대체로 화학적 직관의 문제로 남아 있으며, 이는 근본적으로 진화가 이미 샘플링한 것에 의해 제한되는 수고스러운 과정입니다. 알려진 생물학에 선례가 없는 화학적 변환을 위해 어떻게 기능적인 효소를 체계적으로 design할 수 있을까요?
Generative deep learning models는 단백질 design을 혁신하여, 새로운 결합체의 생성과 사전에 정의된 구조적 모티프의 스캐폴딩을 성공적으로 가능하게 했습니다. 그러나 자연계에 존재하지 않는 반응을 위한 효소의 de novo design은 여전히 대부분 실현되지 않은 상태입니다. 현재의 computational 파이프라인은 두 가지 근본적인 한계에 직면해 있습니다. 첫째, 이들은 활성 부위 잔기나 테오자임의 pre-specified된 고정된 기하학적 배열에 크게 의존합니다. 이는 모티프나 정확한 메커니즘이 알려지지 않은 반응을 배제하며 새로운 활성 부위 기하학의 발견을 방해합니다. 둘째, 실험적으로 검증된 generative 파이프라인은 주로 순차적으로 작동합니다. 이들은 먼저 backbone을 generate한 다음 backbone design에 일치하도록 sequence를 고안합니다 (inverse folding). sequence와 구조가 공동으로 기능을 결정하기 때문에, 이러한 분리된 패러다임은 backbone 형성의 중요한 단계에서 sequence 기반의 objectives를 활용할 수 없으며, 그 반대의 경우도 마찬가지입니다.
우리는 단백질 sequences와 3D 구조를 동시에 de novo로 design하는 일반적인 multimodal 프레임워크인 DISCO (DIffusion for Sequence-structure CO-design)를 소개합니다. DISCO designs는 pre-defined된 잔기 모티프 없이 임의의 생체 분자에 conditioned되고 함께 폴딩될 수 있습니다. 새로운 multimodal 파인만-카츠(Feynman-Kac) corrector 방법은 DISCO가 sequence와 구조 objectives를 함께 최적화할 수 있도록 합니다. DISCO는 세밀한 특성 제어를 통해 다양한 생체 분자 표적에 대한 결합체를 generate하는 데 있어 인실리코(in silico)에서 state-of-the-art 결과를 달성합니다. 실험적으로 자연계에 존재하지 않는 촉매 작용에 적용되었을 때, DISCO는 새로운 활성 부위를 가진 다양하고 진화 가능한 카르벤 전달효소를 generate합니다. inverse-folding은 필요하지 않습니다. 이러한 효소는 지향성 진화의 일반적인 출발점을 능가하는 활성을 가지며, 선택된 경우에는 광범위하게 진화된 변이체를 능가하기도 합니다. 궁극적으로 DISCO는 생물학에 이전에 알려지지 않은 생체 촉매 및 단백질 모티프의 발견을 가능하게 하여, DNA로 암호화된 화학적 반응성의 검색 가능한 공간을 확장합니다.
논문 Introduction 요약 노트 (AI 연구자 대상)
1. 기존 Generative Models의 한계 (Problem Statement) 자연계에 존재하지 않는 화학 반응을 위한 효소의 de novo design은 다음 두 가지 근본적인 한계로 인해 기존 computational 파이프라인에서 구현이 어려웠습니다.
- 고정된 기하학적 배열 의존성: 기존 모델은 pre-specified된 활성 부위 잔기(active-site residues)나 테오자임의 기하학적 배열에 크게 의존합니다. 이는 메커니즘이 알려지지 않은 반응에 적용할 수 없으며, novel active-site geometry의 발견을 원천적으로 차단합니다.
- Inverse Folding 패러다임의 구조적 결함: 기존 파이프라인은 backbone을 먼저 generate한 후, 이에 맞춰 sequence를 추론하는 순차적(decoupled) 방식을 사용합니다. 단백질 기능은 sequence와 structure의 공동 상호작용으로 결정됨에도 불구하고, 이 방식은 backbone 형성 단계에서 sequence 기반의 objectives를 전혀 활용할 수 없습니다.
2. 제안 방법론: DISCO (DIffusion for Sequence-structure CO-design) DISCO는 단백질의 sequences와 3D 구조를 동시에 de novo로 design하는 general multimodal 프레임워크입니다.
- Multimodal Co-design: Inverse-folding 단계를 완전히 배제하고, sequence와 structure를 동시에 생성합니다.
- 제한 없는 Condition 부여: Pre-defined된 잔기 모티프 없이도 임의의 생체 분자에 conditioned되어 함께 폴딩(co-folding)될 수 있습니다.
- 핵심 기술 (Multimodal Feynman-Kac corrector): 이 새로운 corrector 기법을 도입함으로써 DISCO는 sequence objectives와 structure objectives를 생성 과정에서 동시에 결합하고 최적화할 수 있습니다.
3. 핵심 성과 및 의의 (Contributions & Results)
- In silico 성능: 다양한 생체 분자 표적에 대해 세밀한 특성 제어가 가능한 binder를 generate하여 state-of-the-art를 달성했습니다.
- 실험적 증명 (De novo 촉매 생성): 자연계에 존재하지 않는 카르벤 전달효소(carbene transferases)를 실험적으로 generate하는 데 성공했습니다.
- 진화적 우수성: 생성된 효소는 지향성 진화(directed evolution)에 사용되는 일반적인 초기 단백질보다 뛰어난 활성을 보이며, 특정 조건에서는 이미 광범위하게 진화된 변이체의 성능을 상회합니다. DNA로 암호화할 수 있는 화학적 반응성의 탐색 공간을 크게 확장했습니다.
쉬운 설명 :
이 논문의 Introduction은 **"완전히 새로운 화학 반응을 일으키는 맞춤형 단백질(효소)을 AI로 어떻게 만들어낼 것인가?"**라는 질문에 대한 답을 제시하고 있습니다.
기존의 AI 단백질 생성 모델들은 건축에 비유하자면 '철골(backbone)'을 먼저 다 세운 뒤에 '시멘트(sequence)'를 바르는 방식이었습니다. 게다가 이미 설계도가 있는 특정 형태의 방(pre-specified된 활성 부위)만 만들 수 있었죠. 이러다 보니 자연계에 아예 존재하지 않는 전혀 새로운 형태의 건물을 짓는 데는 한계가 있었습니다. 철골을 세울 때 시멘트의 특성을 고려하지 못하는 문제도 있었고요.
이 논문에서 만든 DISCO라는 모델은 철골과 시멘트를 동시에(multimodal co-design) 쌓아 올립니다. 어떤 화학 물질을 주면서 "여기에 딱 맞는 효소를 만들어줘"라고 하면, 미리 정해진 틀 없이 그 물질의 모양에 맞춰 구조와 성분을 알아서 동시에 깎아냅니다. 이것을 가능하게 한 기술이 Multimodal Feynman-Kac corrector라는 새로운 수학적 최적화 방법입니다.
결과적으로 이 AI는 자연계에 없던 새로운 효소를 스스로 디자인해 냈고, 이 AI가 만든 효소는 인간이 기존에 실험실에서 힘들게 개량해 오던 효소들보다 더 뛰어난 성능을 발휘했다는 내용입니다.
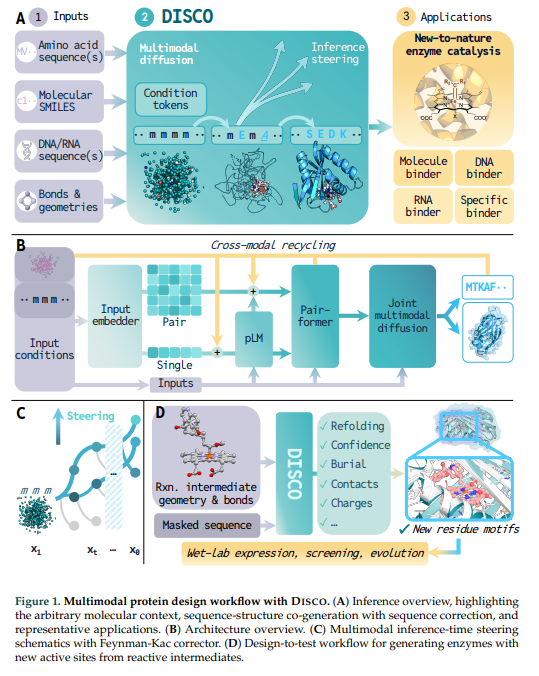
A. Inference overview (추론의 전반적인 흐름)
DISCO가 어떤 입력을 받아 어떻게 작동하고, 어디에 쓰이는지를 직관적으로 보여줍니다.
- Inputs (입력의 다양성): 아미노산 서열, 화학 분자 구조(Molecular SMILES), DNA/RNA 서열, 결합 및 기하학적 정보 등 임의의 분자적 맥락(arbitrary molecular context)을 조건으로 입력받을 수 있습니다.
- DISCO (핵심 생성 엔진): 입력된 Condition tokens를 바탕으로 Multimodal diffusion 과정을 거칩니다. 그림 중앙을 보면 노이즈 상태의 뭉개진 구조와 마스킹된 서열(m m m)이 시간이 지남에 따라 점진적으로 뚜렷한 3D 구조와 명확한 아미노산 서열(S E D K)로 **동시에 생성(co-generation)**되는 것을 볼 수 있습니다. 이 과정에서 Inference steering 기법이 개입하여 원하는 목적에 맞게 생성을 유도합니다.
- Applications (응용 분야): 생성된 단백질은 자연계에 존재하지 않던 새로운 효소 촉매(New-to-nature enzyme catalysis) 역할을 하거나, 특정 분자, DNA, RNA에 특이적으로 결합하는 binder로 활용됩니다.
B. Architecture overview (모델 아키텍처 내부 구조)
DISCO를 구성하는 신경망의 데이터 흐름을 보여줍니다.
- Input embedder: 입력된 다양한 조건들은 이곳을 거쳐 Single 및 Pair 형태의 representations(표현)으로 변환됩니다.
- pLM & Pairformer: 이 정보들은 사전 학습된 단백질 언어 모델(pLM)을 통해 서열 정보를 주입받고, Pairformer 스택을 통과하며 문맥화(contextualize)됩니다.
- Joint multimodal diffusion: 최종적으로 이 모듈이 원자 수준의 주의(atom-level attention)를 기반으로 서열(MTKAF...)과 3D 구조의 좌표를 동시에 예측합니다.
- Cross-modal recycling (핵심 메커니즘): 그림 상단의 노란색 화살표 사이클입니다. 이전 단계에서 예측된 서열 및 구조 정보와 현재 노이즈가 낀 상태 정보가 다시 다음 생성 단계의 조건으로 순환 투입됩니다. 이를 통해 서열은 새롭게 형성되는 구조적 특징을 반영하고, 구조는 진화하는 서열 정체성에 적응하게 됩니다.
C. Multimodal inference-time steering (추론 단계의 방향 조절)
Introduction에서 언급된 Feynman-Kac corrector가 적용되는 과정을 추상적으로 표현한 다이어그램입니다.
- 완전한 노이즈 상태($x_1$)에서 깨끗한 단백질 상태($x_0$)로 향하는 diffusion trajectory(궤적) 중간($x_t$)에 **Steering(방향 조절)**이 개입하는 모습을 보여줍니다. 이를 통해 모델은 서열 objectives와 구조 objectives를 동시에 만족하는 방향으로 최적화 경로를 틀 수 있습니다.
D. Design-to-test workflow (설계부터 실제 실험까지의 파이프라인)
AI 모델의 결과물이 실제 생물학 실험실(Wet-Lab)의 검증으로 이어지는 파이프라인입니다.
- 새로운 반응 중간체의 기하학적 정보와 마스킹된 서열을 DISCO에 입력합니다.
- DISCO는 내부적으로 접힘 가능성(Refolding), 모델의 신뢰도(Confidence), 매몰도(Burial), 접촉(Contacts), 전하(Charges) 등 다양한 지표를 평가합니다.
- 이러한 필터링을 통과하여 새로운 잔기 모티프(New residue motifs)를 가진 유망한 효소 디자인이 도출되면, 이를 Wet-Lab으로 가져가 실제 유전자 발현, 스크리닝, 그리고 지향성 진화(directed evolution) 과정을 거쳐 최종적인 생체 촉매로 완성합니다.
요약하자면:
이 그림은 DISCO가 다양한 입력을 처리하여 서열과 구조를 동시에 깎아내는 신경망 아키텍처(B)를 가지고 있으며, 생성 중간에 궤적을 수정하는 수학적 기법(C)을 활용하여, 최종적으로 실험실에서 당장 쓸 수 있는 완전히 새로운 맞춤형 효소(A, D)를 뱉어내는 완결된 파이프라인임을 보여주고 있습니다.
Multimodal protein generation with DISCO
DISCO는 이산적인 sequences와 3D structures를 단일한 generative process를 통해 denoised되는 joint distribution으로 models합니다 (Fig. 1). 우리는 sequences를 위한 masked discrete diffusion process를 사용하고, 병렬적으로 3D 원자 좌표를 위한 continuous diffusion을 활용하는 단일 deep neural network를 training함으로써 이를 달성합니다. training 중 modality별로 독립적으로 noise를 sampling함으로써, 오직 unimodal diffusion losses만을 사용하여 joint reverse process를 증명 가능하게 학습할 수 있습니다: 이는 좌표를 위한 표준 denoising score matching loss와 sequences를 위한 masked diffusion language model loss입니다. 우리는 특별한 filtering 없이 Protein Data Bank의 data만을 사용하여 DISCO를 trained했으며, 이를 통해 folding algorithms이 성공적으로 predict할 수 있는 designable structures에 대해서만 models가 trained될 때 발생하는 어떠한 selection biases도 방지했습니다. DISCO의 architecture는 모든 input modalities의 single 및 pair representations를 생성하는 input embedder, 부분적인 sequences를 encodes하는 frozen protein language model (pLM), 이러한 representations를 contextualizes하는 Pairformer stack, 그리고 sequence logits와 backbone의 denoised coordinates를 모두 predicts하는 atom-level attention 기반의 joint diffusion module로 구성됩니다 (Fig. 1B). 우리는 architectural constraints가 아닌 data augmentation을 통해 SE(3) symmetries를 부드럽게 inject합니다.
우리의 접근 방식의 핵심은 각 generation step을 네 가지 구별되는 encodings에 conditions하는 cross-modal recycling mechanism입니다: 이는 model의 현재 predicted clean sequence $\hat{x}^{\text{seq}}_0$ 및 structure $\hat{x}^{\text{struct}}_0$, 그리고 현재 noised sequence $x^{\text{seq}}_\tau$ 및 structure $x^{\text{struct}}_\tau$ 입니다. 우리는 structure encoder를 train하고 pLM을 활용하여 sequence information을 inject합니다. 이러한 bidirectional conditioning은 sequence predictions가 새롭게 나타나는 structural features에 의해 정보를 얻도록 보장하는 동시에, structural predictions가 점진적으로 변화하는 sequence identity에 적응하도록 합니다.
DISCO의 inference strategy는 generation quality에 결정적인 것으로 판명되었습니다 (Fig. 2B, Fig. S4). revision 없이 반복적으로 tokens를 unmasking하는 standard masked diffusion은 생성된 geometries로 fold되지 못하는 sequences를 산출합니다. sequence inference 동안 self-correction을 활성화하고, trajectory 초기에 과도하게 확신하는 tokens에 대해 아미노산 distribution을 smooth하게 만드는 새로운 sequence temperature mechanism을 도입함으로써, 우리는 co-designability를 획기적으로 향상시킵니다. 이를 noisy guidance와 결합함으로써 DISCO는 sequence-structure co-design에서 state-of-the-art performance를 달성할 수 있습니다. unconditional monomer design에서, generated sequences의 약 90%가 ESMFold를 사용했을 때 designed backbone structures의 2 Å RMSD 이내로 refold됩니다 (Fig. 2A, Fig. S5, Table S4). baselines와 비교할 때, DISCO는 co-designability를 희생하지 않으면서도 가장 높은 sequence and structure diversity와 novelty를 달성합니다 (Fig. 2A).
Multimodal protein generation with DISCO 요약 노트 (AI 연구자 대상)
1. Unified Generative Process 및 Training 전략
- Joint Distribution 모델링: 이산적인 sequence(masked discrete diffusion)와 연속적인 3D structure(continuous diffusion)를 단일 네트워크에서 병렬로 처리합니다.
- Loss Function 설계: 학습 중 modality별로 noise 를 독립적으로 sampling합니다. 이를 통해 복잡한 multimodal loss 없이, unimodal diffusion losses(좌표에 대한 denoising score matching, 서열에 대한 masked diffusion LM loss)만으로 joint reverse process를 수학적으로 완벽하게 학습할 수 있음을 증명했습니다.
- Unbiased Dataset: "Designable" structure에 편향되는 것을 막기 위해 별도의 필터링 없이 날것의 PDB(Protein Data Bank) 데이터를 그대로 사용하여 학습했습니다.
2. Model Architecture
- 구성 요소: Input embedder $\rightarrow$ frozen pLM(부분 서열 인코딩) $\rightarrow$ Pairformer stack(문맥화) $\rightarrow$ Joint diffusion module(atom-level attention 기반) 순서로 흐릅니다.
- SE(3) Equivariance: 엄격한 architectural constraints로 SE(3) symmetries를 강제하지 않고, data augmentation을 통해 부드럽게(soft) 주입하여 모델의 유연성을 높였습니다.
3. 핵심 혁신 요소 1: Cross-modal recycling
- Bidirectional Conditioning: 매 generation step마다 4가지 상태(예측된 깔끔한 $\hat{x}^{seq}_0$와 $\hat{x}^{struct}_0$, 현재 노이즈가 낀 $x^{seq}_\tau$와 $x^{struct}_\tau$)를 조건으로 부여합니다.
- 이를 통해 서열 예측은 뼈대의 3D 구조적 특징을 반영하고, 3D 구조 예측은 점진적으로 결정되는 서열 정체성에 실시간으로 적응합니다.
4. 핵심 혁신 요소 2: Inference Strategy 최적화
- 문제 제기: 기존의 standard masked diffusion은 서열과 구조가 따로 노는 현상(생성된 geometry로 폴딩되지 않음)이 발생했습니다.
- 해결책 (Sequence Temperature Mechanism): Trajectory 초기 단계에서 모델이 특정 token(아미노산)에 대해 과도하게 확신하는 것을 막기 위해 분포를 평탄화(smooth)하고, self-correction을 도입했습니다.
- 결과 (SOTA 달성): Noisy guidance와 결합하여 co-designability를 비약적으로 상승시켰습니다. Unconditional monomer 생성 시 ESMFold 기준 약 90%가 2 Å RMSD 이내로 정확히 refolding되며, 다양성과 참신함 모두 baseline을 압도합니다.
쉬운 설명 :
이 섹션은 DISCO 모델이 구체적으로 어떻게 단백질의 '모양(3D 구조)'과 '재질(아미노산 서열)'을 동시에 만들어내는지 그 내부 기술을 설명합니다.
건물을 지을 때 뼈대(구조)를 먼저 다 세우고 시멘트(서열)를 바르는 게 기존 방식이었다면, DISCO는 뼈대를 올리면서 동시에 알맞은 시멘트를 배합하는 마법 같은 방식을 씁니다. 이때 두 작업자가 서로 소통하지 않으면 건물이 무너지겠죠? 그래서 DISCO는 Cross-modal recycling이라는 기술을 써서 뼈대 담당 AI와 시멘트 담당 AI가 매 순간 서로 진행 상황을 확인하며 맞춰가도록(Bidirectional conditioning) 설계되었습니다.
또한, 초반부터 "이 자리는 무조건 철근이야!"라고 너무 일찍 확정 지어버리면 나중에 구조가 꼬일 수 있습니다. 그래서 Sequence Temperature Mechanism이라는 장치를 도입해, 작업 초기에는 판단을 유보하고 부드럽게 넘어가도록 만들어 최종 완성도(Co-designability)를 극도로 끌어올렸습니다. 그 결과, AI가 상상으로 만들어낸 단백질 설계도 중 90% 이상이 실제 시뮬레이션에서도 완벽하게 제 모양을 갖추는 놀라운 성능을 달성하게 되었습니다.
Conditioning on arbitrary biomolecular contexts
DISCO는 small molecules, metallocofactors, reactive intermediates, 그리고 nucleic acids를 포함한 임의의 biomolecular contexts에 유연하게 conditions를 부여합니다. 단백질이 아닌 분자들은 화학적 정체성을 나타내는 이산적인 tokens와 그에 상응하는 원자 coordinates를 통해 단백질과 동일하게 표현됩니다. 이 model은 small molecules 및 metal cofactors에서부터 reactive intermediates와 nucleic acids에 이르는 targets의 coordinates를 denoising하는 동시에 protein sequences를 unmasking합니다. 이러한 contexts는 generative trajectory 전반에 걸쳐 designed protein과 함께 co-fold됩니다. DISCO는 단백질을 designing하는 동안 conditioning biomolecules의 coordinates를 조정하여, 분자 상호작용에 의해 유도되는 구조적 변화를 포착하고 고정된 pre-defined atomic scaffolds를 넘어 generation을 확장합니다.
이러한 능력을 benchmark하기 위해, 우리는 catalysis, pharmaceuticals, luminescence 및 sensing 분야의 응용을 포괄하는 179개의 자연 및 비자연 ligands 라이브러리인 새로운 benchmark, STUDIO-179를 설계했습니다. 이 라이브러리는 단단한 분자 (예: 오염 물질인 테트라클로로디벤조다이옥신), 크거나 유연한 분자 (예: 조효소 CoQ10), 그리고 금속/금속 클러스터 (예: [4Fe-4S])를 포함합니다. 각 ligand에 대해, 우리는 구조적으로 다양하고 co-designable한 생성된 designs의 비율을 정량화하며, co-designable을 Chai-1을 사용하여 refolding했을 때 단백질 backbone과 모든 ligand의 중심점이 $RMSD < 2 \text{\AA}$를 가지는 경우로 정의합니다. baselines와 비교하여, DISCO는 평가된 179개의 ligands 중 178개에 대해 가장 높은 비율의 다양하고 co-designable한 complexes를 generates합니다. 우리의 접근법은 동시에 여러 개의 서로 다른 ligands와 interface하도록 단백질을 designing하는 것과 같은 multi-context conditioning을 자연스럽게 지원하며, 이를 통해 다시 한번 고품질의 complexes를 산출합니다.
small molecules를 넘어서, 우리는 macromolecular interfaces에 대한 DISCO의 performance를 평가했습니다. DISCO는 기존 models를 능가하며, sequence-specific DNA 및 RNA sequences 모두에 결합할 것으로 예측되는 co-designable 단백질을 성공적으로 generated했습니다. DISCO 내부의 frozen pLM은 multimeric complexes에 대해 trained되지 않았기 때문에, 우리는 현재의 평가를 non-protein-binding tasks로 제한했다는 점에 유의하십시오.
Conditioning on arbitrary biomolecular contexts 요약 노트 (AI 연구자 대상)
1. Universal Context Representation (범용적 조건 표현)
- 통일된 데이터 처리: Small molecules, metal cofactors, 반응 중간체(reactive intermediates), DNA/RNA 등 임의의 생체 분자를 단백질과 완벽하게 동일한 포맷(이산적인 tokens 및 해당 원자 coordinates)으로 표현하여 입력값으로 받습니다.
2. Dynamic Co-folding (동적 공동 폴딩 & 구조적 변형 포착)
- 핵심 차별점: 기존 모델들처럼 타겟 분자를 '고정된 뼈대(pre-defined fixed scaffolds)'로 두고 단백질을 끼워 맞추는 방식이 아닙니다.
- 상호작용 반영: 단백질의 sequences를 unmasking하고 coordinates를 denoising하는 생성 과정 내내, 조건으로 부여된 타겟 분자의 좌표도 **함께 미세 조정(adjust)**합니다. 이를 통해 분자 간 상호작용으로 인해 발생하는 유동적인 구조적 변화(conformational changes)를 모델이 스스로 포착하고 반영할 수 있습니다.
3. 새로운 벤치마크 (STUDIO-179) 제안 및 SOTA 달성
- 평가 지표 구축: 촉매, 제약, 센싱 등 다양한 응용 분야를 포괄하는 179종의 리간드(rigid, flexible, 금속 클러스터 등 모두 포함) 라이브러리인 'STUDIO-179'를 새롭게 설계하여 평가에 활용했습니다.
- 압도적 성능: Chai-1을 이용한 refolding 시 단백질 backbone과 모든 리간드의 중심점(centroid)이 $RMSD < 2 \text{\AA}$를 만족하는 것을 'co-designable'하다고 정의했을 때, 179개 중 178개 리간드에서 베이스라인을 압도하며 가장 높은 생성 비율(SOTA)을 기록했습니다.
- Multi-context 지원: 여러 개의 서로 다른 타겟 리간드가 동시에 존재하는 복잡한 환경(multi-context conditioning)에서도 고품질의 복합체를 설계해 냈습니다.
4. Macromolecular Interfaces (거대 분자) 확장성
- Small molecules를 넘어, 특정 서열의 DNA 및 RNA와 결합하는 단백질 설계에서도 기존 모델들의 성능을 뛰어넘었습니다.
- 한계점 노트: DISCO 내부의 frozen pLM이 다량체 복합체(multimeric complexes)에 대해 학습되지 않았기 때문에, 이번 연구에서는 단백질-단백질 결합(protein-binding) 생성은 평가에서 제외했습니다.
쉬운 설명 :
이 섹션은 DISCO가 **"어떤 종류의 목표물이 주어지든, 그 모양에 완벽하게 딱 맞는 맞춤형 단백질 장갑을 어떻게 짜낼 수 있는지"**를 설명합니다.
과거의 AI 모델들은 목표물을 '절대 움직이지 않는 단단한 돌멩이' 취급을 하고 그 주위에 단백질을 조립했습니다. 하지만 실제 생물학적 환경에서는 두 물질이 만날 때 서로 영향을 주고받으며 모양이 미세하게 변합니다. DISCO는 장갑(단백질)을 짜는 과정에서, 안에 들어있는 손(목표물 분자)의 관절이 살짝 굽혀지거나 펴지는 것(Conformational changes)까지 동시에 시뮬레이션하면서 가장 편안하게 밀착되는 형태를 찾아냅니다.
게다가 이 목표물이 작은 화학물질이든, 금속 덩어리이든, 심지어 거대한 DNA 사슬이든 가리지 않고 똑같은 방식으로 처리할 수 있는 엄청난 범용성을 갖췄습니다. 연구진이 일부러 까다로운 179개의 목표물(STUDIO-179)을 던져주며 테스트해 보았는데, 무려 178개에서 기존 AI들을 압도하며 완벽하게 들어맞는 단백질을 생성해 내는 데 성공했습니다. 한마디로 **"목표물의 종류나 크기에 상관없이, 살아 숨 쉬는 듯한 유연한 맞춤 제작이 가능한 만능 AI"**라는 것을 증명한 파트입니다.