

AI바라기의 인공지능
protein : 논문 리뷰 : MarS-FM: Generative Modeling of Molecular Dynamics via Markov State Models 본문
protein : 논문 리뷰 : MarS-FM: Generative Modeling of Molecular Dynamics via Markov State Models
AI바라기 2026. 4. 10. 20:32
용어 설명
- MD (Molecular Dynamics): 분자의 원자 수준 움직임을 뉴턴 역학으로 시뮬레이션하는 연산 기법. 매우 긴 시간이 소요되는 것이 단점입니다.
- MD-Emu (MD Emulator): 고정된 시간 간격(lag time, tau)에 따른 MD 프레임 간의 단기적인 물리적 전이(transition)를 모사하여 연속적인 궤적을 생성하도록 학습된 생성 모델 클래스입니다.
- MSM (Markov State Model): 방대한 연속적 MD 궤적을 의미 있는 몇 개의 이산적인 준안정 상태(metastable states)로 군집화(coarse-graining)하고, 이 상태들 간의 전이 확률을 마르코프 체인으로 나타낸 모델입니다.
- MSM-Emu (MSM Emulator): 이 논문에서 새롭게 제안한 생성 모델 클래스로, 시간순의 프레임 전이가 아닌 MSM이 정의하는 거시적인 '상태 간의 전이(state-to-state transition)'를 샘플링하도록 학습됩니다.
- MarS-FM (Markov Space Flow Matching): 논문에서 제안한 MSM-Emu 클래스의 대표 아키텍처. Flow Matching 기법을 사용하여 추출된 출발 상태와 도착 상태 사이를 부드럽게 보간(interpolation)하여 구조를 생성합니다.
- TICA (Time-lagged Independent Component Analysis): 시간 지연 자기상관성을 최대화하여 분자 동역학 데이터의 차원을 축소하는 기법입니다.
- Autoregressive sampling: 이전 시점에서 생성된 프레임을 다음 생성의 입력 조건으로 계속 연쇄적으로 사용하는 샘플링 방식입니다. 긴 궤적 생성 시 오류 누적(compounding error)을 일으킵니다.
Purpose of the Paper
- 기존 연구의 한계 (데이터 불균형 및 탐색 실패): 기존의 MD-Emus는 고정된 시간 간격의 프레임 변화를 학습합니다. 이로 인해 에너지가 안정된 상태 안에서의 미세하고 무의미한 진동(intra-state)에만 학습 데이터가 편중되며, 에너지가 높은 장벽을 넘어야 하는 중요한 구조적 변화(예: 단백질 풀림 현상 등 inter-state)는 학습하지 못하는 치명적인 한계가 있었습니다. 또한 autoregressive sampling에 의존하여 오차가 빠르게 누적됩니다.
- 새로운 문제 정의 및 접근 (시간적 동역학으로부터의 탈피): 본 연구는 모델의 학습 목표를 '미시적인 시간적 동역학(temporal dynamics) 재현'에서 완전히 분리해야 한다고 정의합니다. 대신, 데이터를 사전에 MSM으로 요약한 뒤, 모델이 특정 시간이 아닌 '거시적 상태 간의 변화' 자체를 직접 생성하도록 유도하는 MSM-Emus라는 새로운 생성 패러다임을 제안합니다.
Key Contributions
- 새로운 생성 모델 클래스 MSM-Emus 및 MarS-FM 제안: 기존 MD-Emus와 달리, 사전에 구축된 MSM의 전이 행렬을 기반으로 출발 상태와 도착 상태를 균일하게 먼저 고른 뒤, 해당 상태에 속한 프레임들을 Flow Matching으로 연결하여 학습하는 구조를 확립했습니다.
- 참신성 (학습 신호의 혁신적 전환): 단순히 모델 architecture를 키우는 것이 아니라, 학습 쌍(training pairs)을 구성하는 방식 자체를 '고정 시간 간격의 프레임'에서 'MSM 상태 기반의 무작위 프레임'으로 바꾸는 역발상을 통해 고질적인 데이터 불균형과 희소성 문제를 근본적으로 우회한 점이 매우 참신합니다.
- Fast exploration 및 Autoregressive 의존성 제거: 상태를 기반으로 샘플링하기 때문에 이전 프레임에 얽매이지 않고 여러 프레임을 병렬(parallel) 또는 계층적 트리(hierarchical tree) 형태로 동시 생성할 수 있습니다. 이는 누적 오차를 획기적으로 줄이고 에너지 지형도 탐색 효율을 극대화합니다.
- 하이브리드 샘플링 파이프라인 (MarS-FM + MDGen): 거시적인 상태 탐색은 MarS-FM이 담당하고, 특정 상태 내부에서의 세밀한 시간적 움직임은 기존 MD-Emu가 담당하도록 결합하여 두 방법론의 장점만을 취하는 방식을 제시했습니다.
Experimental Highlights
- 핵심 실험 설정 (Datasets & Generalization Strictness): 4AA Tetrapeptides 및 대규모 단백질 데이터셋인 MD-Cath를 사용했습니다. 특히 모델의 강력한 일반화(generalization) 능력을 입증하기 위해, 테스트 데이터셋은 학습 데이터와 서열 유사성이 20% 이하인 완전히 새로운 단백질(unseen proteins)들로만 구성하는 매우 엄격한 기준을 적용했습니다. 또한 극적인 구조 변화를 보기 위해 단백질이 풀리는 450K 고온 시뮬레이션 데이터를 주력으로 사용했습니다.
- 압도적인 유연성 및 자유 에너지 모사 (SOTA 달성): MD-Cath 테스트 결과, MarS-FM은 기존 SOTA인 MDGen 대비 Global RMSF Pearson correlation에서 0.71 (MDGen 0.49)을 기록하며 분포 모사 능력을 크게 높였습니다. 단백질 접힘 자유 에너지 오차(Delta G fold MAE)에서도 1.05 kcal/mol을 기록하여 베이스라인(1.21~2.14)을 압도했습니다.
- 막대한 계산 속도 향상: 159개 잔기를 가진 단백질의 500ns 분량 궤적을 생성할 때, implicit solvent MD 시뮬레이션은 약 5시간이 걸리지만 MarS-FM은 단 30초 만에 생성을 완료하여 600배 이상의 속도 향상을 증명했습니다.
Limitations and Future Work
- Limitations 1 (초기 구조 의존성): inference 시에 여전히 단백질의 초기 3D 구조 정보가 입력으로 필요합니다. 이는 서열만 알고 있는 새로운 단백질에 바로 적용하기 어렵게 만듭니다.
- Limitations 2 (미세 동역학 재현의 한계): MSM-Emus는 거시적 상태 탐색과 열역학적 일치도에 초점이 맞춰져 있어, 아주 짧은 시간 단위의 정밀한 프레임 간 순서(temporal ordering)나 국소적인 동역학은 기존 MD-Emus만큼 완벽하게 재현하지 못합니다.
- Future Work:
- 최신 sequence-to-structure 생성 모델들과 결합하여, 3D 구조 입력 없이 아미노산 서열(sequence)만으로도 초기 구조와 동역학을 모두 생성할 수 있도록 파이프라인을 확장할 계획입니다. 이를 통해 한계를 극복하고 범용성을 높일 수 있습니다.
- 단일 단백질을 넘어 단백질-리간드 복합체(complexes)나 무기물 소재 시스템으로 프레임워크를 확장하여 신약 개발 등 실질적인 산업 적용도를 높일 예정입니다.
- MarS-FM으로 에너지 지형의 다양한 상태를 먼저 짚어낸 뒤, 거기서부터 실제 짧은 MD 시뮬레이션을 병렬로 시작하게 하여 정확도와 속도를 동시에 잡는 융합 연구를 제안합니다.
Overall Summary
이 논문은 고정된 시간 간격에 얽매여 비효율적인 탐색을 하던 기존 분자 동역학 생성 모델의 한계를 깨고, 마르코프 상태 모델(MSM) 기반의 거시적 전이를 직접 학습하는 완전히 새로운 생성 패러다임인 MarS-FM을 제안했습니다. 서열이 전혀 다른 미지의 단백질에 대해서도 극적인 구조 변화를 정확하고 다양하게 생성해 냈으며, 실제 시뮬레이션 대비 600배 이상의 획기적인 속도 향상을 입증했습니다. 이는 단백질의 장기적인 열역학적 앙상블을 계산 효율적으로 추론할 수 있게 하여, 향후 AI 기반 신약 발굴 및 분자 설계 파이프라인의 핵심 가속 엔진으로 활용될 수 있는 매우 중요한 학술적, 실용적 의의를 지닙니다.
쉬운 설명
기존의 AI 모델(MD-Emu)이 비디오를 흉내 내는 방식을 보면, "1초 뒤의 다음 픽셀 장면"을 연속적으로 예측하려고 했습니다. 그러다 보니 주인공이 제자리에 가만히 서서 숨만 쉬는 지루한 장면만 잔뜩 학습하게 되고, 정작 건물에서 뛰어내리는 '큰 액션 씬(거시적 구조 변화)'은 거의 본 적이 없어 만들어내지 못했습니다. 게다가 1초 단위로 계속 예측을 이어나가다 보니 나중에는 픽셀이 뭉개지는 에러가 쌓였습니다.
반면 이 논문이 만든 MarS-FM은 비디오를 프레임 단위로 보지 않습니다. 비디오의 핵심 챕터(MSM 상태)들을 먼저 요약해 둔 다음, "방 안에 있던 주인공(상태 A)이 다음 챕터에서는 옥상(상태 B)에 있다"는 굵직한 스토리 흐름 자체를 뽑아내어 다이렉트로 장면을 스케치(Flow Matching)합니다. 쓸데없는 1초 단위의 시간 제약에 얽매이지 않기 때문에 에러가 쌓이지도 않고, 단백질이 완전히 접히거나 풀리는 스펙터클한 액션 씬들을 기존 방식보다 수백 배 빠르고 정확하게 쑥쑥 만들어낼 수 있는 것입니다.
인풋 3d 원자 구조, 서열, time, 노이즈
아웃풋 타겟 3d 원자 구조로 가기 위한 방향 (v 프레드)
인퍼런스 ode로 해서 3d 좌표 생성
가우시안 노이즈~ 타겟 타입 구조 까지의 타입에 따른 데이터가 있고
입력 타입 구조와 서열을 참고해서 학습방법을 배우는데
인퍼런스에서도 초기3d 구조 랑 노이즈, 시간 아랑 서열 가지고 복원시켜버림
Molecular Dynamics (MD)는 단백질 기능을 탐구하기 위한 강력한 계산 현미경입니다. 그러나 세밀한 통합에 대한 필요성과 생체 분자 이벤트의 긴 시간 척도로 인해 MD는 계산적으로 막대한 비용이 소모됩니다. 이를 해결하기 위해 더 적은 비용으로 surrogate trajectories를 generate하기 위한 여러 generative models가 제안되었습니다. 그러나 이러한 models는 일반적으로 fixed-lag transition density를 learn하며, 이로 인해 training signal이 발생 빈도만 높고 정보가 부족한 transitions에 의해 지배되는 현상이 발생합니다.
우리는 기반이 되는 Markov State Model (MSM)에 의해 정의된 discrete states 간의 transitions를 sample하는 방법을 learn하는 새로운 generative models 클래스인 MSM Emulators를 소개합니다. 우리는 이 클래스를 Markov Space Flow Matching (MarS-FM)으로 instantiate하며, 이 모델의 sampling은 implicit- or explicit-solvent MD 시뮬레이션에 비해 두 자릿수 이상의 속도 향상을 제공합니다.
우리는 RMSD, radius of gyration, secondary structure content와 같은 structural observables를 통해 MD 통계를 재현하는 MarS-FM의 능력을 benchmark합니다. 우리의 평가는 unfolding 이벤트를 포함하여 화학적 및 구조적 다양성이 큰 단백질 도메인(최대 500 residues)에 걸쳐 수행되며, generalization을 평가하기 위해 training과 test sets 간의 엄격한 sequence dissimilarity를 강제합니다. 모든 metrics에 걸쳐 MarS-FM은 기존 방법들을 상당한 차이로 outperforms 합니다. Code는 https://github.com/valence-labs/mars-fm 에서 이용 가능합니다.
1 Introduction
Deep Learning은 단백질의 3D structures에 대한 빠르고 정확한 prediction을 가능하게 했습니다. 그러나 이러한 methods는 Boltzmann distribution에 의해 지배되는 conformational ensembles를 가지는 단백질의 동적 거동을 capture하지 못합니다. 이러한 dynamics를 연구하기 위한 가장 신뢰할 수 있는 계산 도구는 Molecular Dynamics (MD)로, Newton의 법칙을 통합하여 원자 운동을 시뮬레이션합니다. 긴 MD trajectories는 Boltzmann distribution으로부터의 samples를 제공하고 생체분자 interactions의 메커니즘을 밝혀주며, 이는 신약 개발의 핵심 자산입니다. 그럼에도 불구하고 생물학적 이벤트가 시뮬레이션 timestep보다 훨씬 긴 시간 척도에서 발생하기 때문에 MD는 비용이 많이 듭니다. 이러한 과제는 종종 비물리적인 힘을 통해 dynamics를 가속화하는 다양한 향상된 sampling methods를 촉진했습니다.
최근 연구들은 Boltzmann distribution에서 sample하기 위해 generative flows를 사용하여 MD의 계산적 부담을 피하는 것을 제안했습니다. 핵심 하위 클래스는 고정된 lag time $\tau$와 관련된 transition density를 modeling하여 MD-sampling을 emulate하도록 learn하는 MD Emulators (MD-Emus) 제품군입니다. 즉, MD-Emus는 특정 입력 프레임 $x(t)$를 조건으로 향후 conformations $x(t + \tau)$를 generate하는 models입니다. 추론 시 이러한 methods는 surrogate MD trajectories를 produce하기 위해 자기회귀적으로 적용됩니다. Jing et al. (2024b)은 고정된 간격 $\tau$로 분리된 여러 프레임을 공동으로 generate하도록 model을 training하여 이 접근 방식을 발전시켰습니다. 그러나 고정된 lag time에서 dynamics를 learning하는 것은 주요한 과제를 도입합니다. 짧은 lag times는 달성 가능한 속도 향상을 제한하는 반면, 긴 lag times는 중요한 meta-stable states를 건너뛸 수 있습니다. 보다 근본적으로, training signal은 시뮬레이션 중에 관찰된 발생 빈도는 높지만 정보가 부족한 transitions에 의해 지배되는 반면, exploration을 주도하는 high-barrier transitions는 여전히 과소대표됩니다. 결과적으로 MD-Emus는 드물고 큰 conformational changes를 capture하는 데 어려움을 겪을 수 있습니다.
MD-Emus의 한계는 시간적 dynamic에 관련 없는 고주파 정보가 포함된 MD의 데이터 불균형에서 비롯됩니다. coarse-grained representations를 통해 MD에서 의미 있는 signal을 extract하는 일반적인 전략은 Markov State Models (MSMs)에서 제공합니다. MSMs는 프레임을 discrete states로 cluster하고 Markov chain matrix $T$를 통해 dynamics를 설명합니다. MSMs는 동적 콘텐츠를 압축하면서 $T$에 의해 유도된 equilibrium distribution을 통해 장기 통계의 추정을 보장합니다.
Main Contributions
우리는 MD trajectories로부터 구성된 MSM에 의해 유도된 Markov-chain transition에서 sample하도록 learn하는 새로운 generative models 클래스인 MSM Emulators (MSM-Emus)를 제시합니다. 이러한 관점에서 MSM은 metastable states 간의 long-timescale dynamics의 coarse-grained Markov model을 제공하며, MSM-Emus는 전체 고주파 MD 시계열이 아닌 metastable states 간의 transitions를 learning하는 데 중점을 둡니다.
target transitions가 이제 특정 관찰된 paths가 아닌 state 연결성에 의존하기 때문에, MSM-Emus는 높은 에너지 장벽을 넘는 드문 conformational changes를 learn하기 위한 훨씬 더 높은 training diversity의 이점을 얻습니다. 사실, MSM-Emus는 특정 프레임들 사이가 아니라, 접힘이나 풀림과 같은 큰 conformational changes와 관련된 discrete macroscopic states에 걸쳐 보간(interpolate)하며, 이는 서로 다른 단백질들 사이에서 더 견고하고 일반화 가능한 signal입니다. sampling하는 동안 프레임은 더 쉽게 역상관(decorrelated)되고 자기회귀 호출이 크게 줄어들어 복합 오류 효과를 완화합니다. 우리는 Flow Matching을 사용하여 최적화된 새로운 framework인 Markov Space Flow Matching (MarS-FM)으로 이 클래스를 선보입니다.
Evaluation with chemical and structural diversity
MD-Emus는 보통 작은 펩타이드나 화학적 또는 구조적 다양성이 제한된 datasets에서 평가되었습니다. 이러한 격차를 해결하기 위해 우리는 sequence 길이가 최대 500 residues인 수천 개의 단백질 도메인의 대규모 dataset인 MD-CATH에서 MarS-FM을 평가합니다. 특히 가장 높은 온도의 레플리카를 활용하여 펼쳐짐(unfolding)과 같은 큰 conformational changes를 capture하는 MarS-FM의 능력을 test할 수 있습니다. generalization을 평가하기 위해 최대 민감도를 가진 mmseqs2를 사용하여 test 단백질이 training set의 어떤 단백질과도 20% 이하의 sequence 유사성을 공유하도록 보장하는 프로토콜을 채택합니다. 우리는 RMSD, 회전 반경(radius of gyration), 2차 구조 내용(secondary structure content)을 포함하는 structural observables를 사용하여 MarS-FM을 benchmark하고, 이것이 지속적으로 MD-Emus를 능가함(종종 큰 차이로)을 보여줍니다. 결정적으로, MarS-FM은 MD와 MD-Emus보다 훨씬 더 효율적으로 target distribution을 탐색하여, 샘플 수가 적은 체제(low-sample regimes)에서도 서로 다른 states에 걸쳐 conformations를 generate합니다.
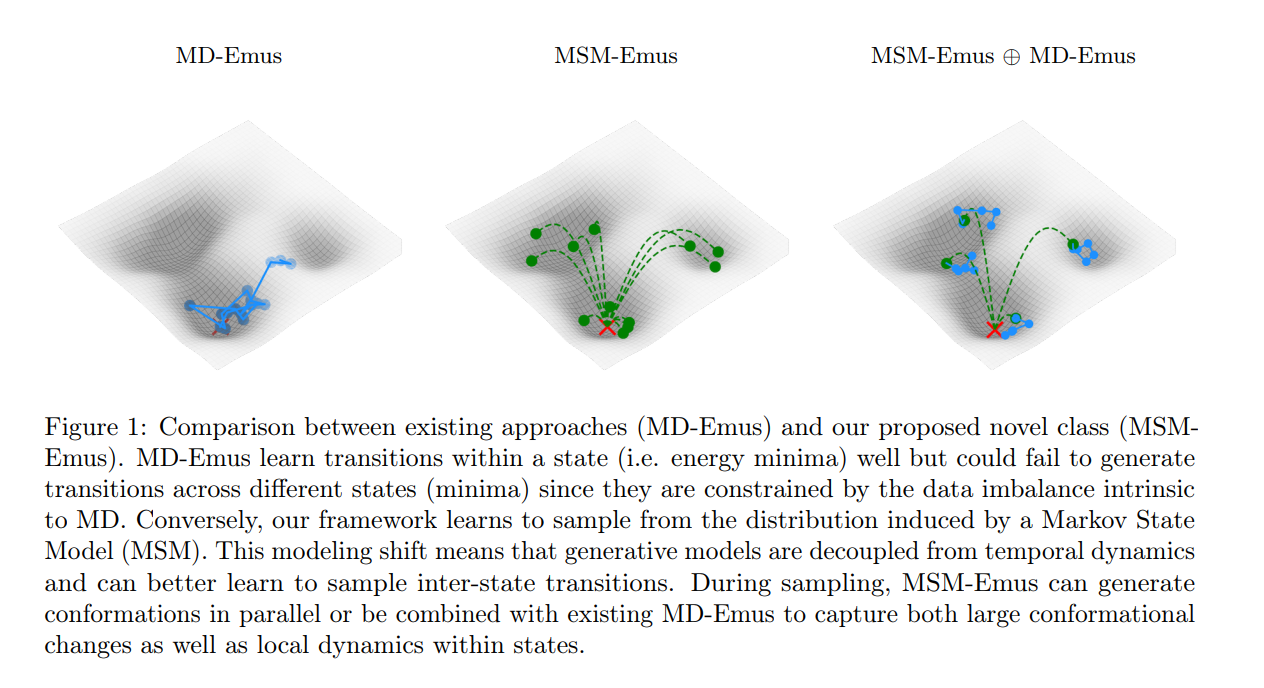
전체적인 구도는 세 개의 도표로 나누어져 있으며, 모두 동일한 물결 모양의 3D 그리드 표면을 배경으로 사용합니다. 이 표면은 고저차가 있는 풍경처럼 보이는데, 아래로 오목한 부분(구덩이)은 단백질이 안정적으로 머무를 수 있는 에너지 최소값(energy minima) 또는 상태(states)를 의미합니다. 가장 왼쪽 아래 구덩이에 표시된 붉은색 X는 시뮬레이션의 출발점입니다.
각 도표의 내용은 다음과 같습니다.
- 1. MD-Emus (가장 왼쪽 도표)
- 시각적 설명: 파란색 점들이 붉은색 X가 있는 구덩이 안에만 모여 있고, 파란색 실선으로 연결되어 있습니다. 이 선들은 출발점이 있는 하나의 에너지 최소값(구덩이) 내에서의 움직임만을 보여주며, 다른 구덩이로 이동하는 모습은 보이지 않습니다.
- 의미 (이전 텍스트와 연결): 기존의 MD-Emus 모델이 고정된 시간 간격($\tau$)으로만 학습하기 때문에, 미세한 떨림(고주파 정보)에만 집중하게 되어 하나의 안정적인 상태 내에서의 움직임(상태 내 전이)은 잘 포착하지만, 다른 상태로 이동하는 큰 변화(상태 간 전이)는 학습하지 못함을 시각적으로 보여줍니다. 캡션에서는 이를 MD 데이터 고유의 데이터 불균형 문제로 설명합니다.
- 2. MSM-Emus (가운데 도표)
- 시각적 설명: 녹색 점들이 네 개의 모든 구덩이(에너지 최소값)에 분포되어 있으며, 이 점들이 녹색 점선으로 연결되어 있습니다. 이 점선은 한 구덩이에서 다른 구덩이로 이동하는 큼직한 움직임을 보여주며, 출발점인 X에서 다른 세 개의 구덩이로 직접 연결되는 여러 경로가 보입니다.
- 의미 (이전 텍스트와 연결): 이 논문에서 제안한 MSM-Emus 모델의 작동 방식을 시각화한 것입니다. 이 모델은 Markov State Model(MSM)에 의해 정의된 상태 간의 transitions를 학습하기 때문에, 프레임 단위의 미세한 움직임이 아니라 서로 다른 안정적인 상태(구덩이) 간의 큰 구조적 변화를 성공적으로 포착하여 샘플링할 수 있음을 보여줍니다.
- 3. MSM-Emus $\oplus$ MD-Emus (가장 오른쪽 도표)
- 시각적 설명: 두 도표의 특징이 결합되어 있습니다. 녹색 점선은 서로 다른 구덩이 간의 큰 이동을 보여주고, 파란색 실선은 각 구덩이 내부의 작은 움직임을 보여줍니다.
- 의미 (이전 텍스트와 연결): MSM-Emus가 독립적으로 작동할 수도 있지만, 기존의 MD-Emus와 결합하여 사용할 수도 있음을 보여줍니다. 이렇게 결합함으로써, 단백질의 크고 중요한 구조적 변화(MSM-Emus 담당)와 상태 내부의 국부적인 세밀한 역학(MD-Emus 담당)을 모두 포착할 수 있다는 장점을 시각적으로 설명합니다.
요약하자면, 이 그림은 MD 데이터의 불균형 문제로 인해 하나의 상태에 갇히는 기존 MD-Emus의 한계와, Markov State Model을 활용하여 상태 간의 큰 전이를 성공적으로 학습하는 제안된 MSM-Emus의 우수성을 명확한 시각적 대비를 통해 설명하고 있습니다.
논문 Introduction 요약 정리노트 (AI 연구자용)
- 배경 및 기존 연구의 한계 (Limitations of MD-Emus)
- Deep Learning은 단백질의 정적인 3D structure prediction에는 능하지만, 동적인 움직임(Boltzmann distribution)을 포착하지 못함. 이를 해결하기 위한 기존의 Molecular Dynamics (MD)는 연산 비용이 지나치게 높음.
- 대안으로 고정된 시간 간격($\tau$)의 transition density를 modeling하여 MD-sampling을 모사하는 생성 모델인 MD-Emus가 등장함.
- Critical Flaw: 고정된 lag time $\tau$로 인해 학습 신호가 발생 빈도만 높고 정보가 없는 미세한 transitions에 편중됨. 결과적으로 단백질의 실질적인 exploration을 이끄는 높은 에너지 장벽의 rare, large conformational changes를 포착하는 데 실패함.
- 핵심 제안 (Main Contributions: MSM-Emus & MarS-FM)
- MD 데이터의 불균형(고주파 노이즈)을 해결하기 위해 Markov State Models (MSMs)의 coarse-grained 방식을 차용하여 새로운 모델 클래스인 MSM-Emus 제안.
- 이 개념을 Flow Matching으로 최적화하여 구현한 MarS-FM (Markov Space Flow Matching) 프레임워크 최초 공개.
- MarS-FM의 작동 원리 및 장점 (Mechanism & Advantages)
- 단순 프레임 단위의 예측이 아니라, MSM에 의해 정의된 metastable states 간의 transitions를 학습함.
- 특정 관찰 경로(observed paths)가 아닌 상태 간의 연결성(state connectivity)을 기반으로 target transitions를 학습하기 때문에, 드물게 일어나는 구조적 변화(folding/unfolding 등)에 대한 training diversity가 비약적으로 상승함.
- 프레임 간 역상관(decorrelated)이 쉬워져 autoregressive 호출 횟수가 대폭 감소하고, 이로 인해 sequence 생성 시 발생하는 복합 오류(compounding error)가 크게 완화됨.
- 가혹한 평가 환경 및 결과 (Rigorous Evaluation)
- 단순한 펩타이드 수준의 기존 평가를 탈피하여, 최대 500 residues 길이를 가진 수천 개의 단백질 도메인 대규모 데이터셋(MD-CATH) 활용.
- Strict Generalization: test set 단백질이 training set과 20% 이하의 서열 유사성만 가지도록 엄격하게 통제(mmseqs2 활용).
- Performance: 구조적 지표(RMSD, radius of gyration 등)에서 기존 MD-Emus를 큰 격차로 outperforms. 특히 적은 샘플(low-sample regimes)만으로도 target distribution을 훨씬 효율적으로 탐색함을 입증.
쉬운 설명 :
이 논문이 풀고자 하는 문제는 "어떻게 하면 단백질의 큼직하고 중요한 움직임만 똑똑하게 AI로 시뮬레이션할 수 있을까?" 입니다.
단백질은 가만히 있는 조각상이 아니라 계속해서 꼬물거리고 접혔다 풀렸다 하는 기계와 같습니다. 기존의 AI 모델(MD-Emus)에게 단백질의 움직임을 예측하라고 시켰더니, AI가 0.001초 단위의 **'의미 없이 미세하게 떨리는 움직임(고주파 정보)'**만 잔뜩 배우느라 정작 중요한 **'단백질이 완전히 펴지거나 접히는 커다란 변화'**는 제대로 배우지 못하는 문제가 발생했습니다. 너무 짧은 간격에만 집착하다 보니 숲을 보지 못하고 나무의 잔떨림만 본 셈입니다.
그래서 연구진은 **MSM-Emus (그리고 이를 구현한 MarS-FM)**라는 새로운 방식을 도입했습니다. 이 방식은 단백질의 움직임을 1프레임 단위의 촘촘한 비디오로 무식하게 다 학습하는 것이 아닙니다. 대신, 단백질이 안정적으로 머무는 '핵심적인 상태(metastable states)'들을 징검다리처럼 정의하고, 이 징검다리 사이를 훌쩍훌쩍 건너뛰는 큼직한 변화(transitions)만 집중적으로 학습하게 만들었습니다.
결과적으로 AI는 쓸데없는 잔떨림을 계산하느라 힘을 빼지 않게 되었습니다. 대신 단백질이 어떻게 크게 형태를 바꾸는지 그 **'핵심 과정'**을 훨씬 빠르고 정확하게 시뮬레이션할 수 있게 되었고, 처음 보는 크고 복잡한 단백질을 대상으로 한 깐깐한 시험에서도 기존 AI들을 압도하는 훌륭한 성적을 거두었습니다.
2 Preliminaries and Setting
온도 $T$에서 열평형(thermal equilibrium) 상태에 있는 $N$개의 원자로 구성된 분자 시스템은 Boltzmann distribution $\mu$에 따라 conformations를 탐색합니다. 즉, $q \sim \mu(q) \propto \exp(-H(q)/(k_BT))$이며, 여기서 $q = (x, \xi) \in X$는 시스템의 위치와 속도를, $H$는 Hamiltonian, 그리고 $k_B$는 Boltzmann 상수를 나타냅니다.
우리는 단백질에 초점을 맞추며, 국소적인 펼쳐짐(local unfolding)이나 숨겨진 포켓 형성(cryptic pocket formation)과 같은 움직임은 구조적 변동(structural fluctuations)에서 비롯됩니다. 이러한 conformational changes를 식별하고 그 빈도(자유 에너지)를 정량화하기 위해, 우리는 Boltzmann distribution $\mu$에서 structures를 sample하는 것을 목표로 합니다. 이는 기댓값 $\mathbb{E}_{x \sim \mu}[\phi(x)]$를 통해 위상 공간 $X$에 정의된 함수인 observables $\phi$의 측정을 가능하게 합니다.
$\mu$로부터 직접 sampling하는 것은 까다롭기 때문에 널리 쓰이는 대안은 Molecular Dynamics (MD)를 사용하는 것입니다. MD는 $\mu$에 대해 에르고딕(ergodic)하도록 설계된 연속 시간 Markov process를 시뮬레이션합니다. 즉, 어떤 observable에 대해서든 긴 trajectory에 걸친 시간 평균은 $\mu$ 하에서의 기댓값에 근사합니다. MD의 일반적인 공식화는 Langevin dynamic을 통합니다. potential energy $U$를 가진 시스템이 주어졌을 때, 질량 $m_i$를 가진 원자 $i$의 위치와 속도는 다음과 같이 업데이트됩니다.
여기서 $\gamma$와 $\sigma$는 마찰(friction)과 열 노이즈(thermal noise)를 제어합니다. 실제로는 운동 성분을 버리고 위치의 trajectories $t \mapsto x(t)$에 초점을 맞춥니다.
MD는 강력한 도구이지만 그 계산 비용이 주요 병목 현상입니다. 생물학적으로 관련된 많은 이벤트는 밀리초 시간 척도에서 발생하는 반면, Langevin dynamics는 펨토초 수준의 시간 단계로 통합해야 하므로 긴 시뮬레이션을 수행하는 데 엄청난 비용이 듭니다. 결정적으로, 단백질의 meta-stable states는 종종 높은 에너지 장벽으로 분리되어 있어, MD 시뮬레이션이 깊은 local minima에 갇혀 시간을 낭비하게 만듭니다.
Problem Formulation
초기 단백질 conformation $x(0)$가 주어지면, MD는 수식을 통합하고 일정한 간격으로 프레임을 저장하여 structures의 앙상블을 produce합니다. MD는 비용이 많이 들기 때문에, 우리는 훨씬 적은 비용으로 대리 분포(surrogate distributions)를 sample하기 위해 generative models를 사용하는 것을 목표로 합니다. 여러 단백질에 걸친 generalization을 가능하게 하기 위해, 여러 sequences에서 사용 가능한 MD trajectories를 사용하여 단일 model을 training합니다.
추론(inference) 시에는 이전에 본 적 없는 단백질의 sequence와 입력 conformation을 제공하며, 목표는 일련의 물리적 observables $\phi$와 관련하여 MD trajectories와 통계적으로 구별할 수 없는 samples를 generate하는 것입니다. 즉, model은 연구자들이 관심을 갖는 구조적 또는 열역학적 양에 대한 분포와 일치해야 합니다. 다음으로 고정된 transition density에서 sampling하여 이 문제를 해결하려고 시도하는 기존 접근 방식들을 검토하고 그 한계점을 논의합니다.
논문 2 Preliminaries and Setting 요약 정리노트 (AI 연구자용)
- 궁극적 목표 (Objective)
- 단백질의 구조적 변동(숨겨진 포켓 형성, 국소적 풀림 등)을 이해하기 위해 **Boltzmann distribution $\mu$**에서 구조를 샘플링하는 것.
- 이를 통해 위상 공간에 정의된 물리적 관측값(observables) $\phi$의 기댓값 $\mathbb{E}_{x \sim \mu}[\phi(x)]$을 계산하는 것이 핵심 목표.
- 기존 방법론(MD)의 구조적 한계 (The Bottleneck of MD)
- Langevin Dynamics 기반의 한계: MD는 $\mu$를 에르고딕(ergodic)하게 탐색하지만, 펨토초(fs) 단위의 연산으로 밀리초(ms) 단위의 생물학적 이벤트를 시뮬레이션해야 하므로 연산 비용이 극도로 높음.
- Local Minima Trapping: 단백질의 준안정 상태(meta-stable states)는 높은 에너지 장벽(high-energy barriers)으로 막혀 있음. MD는 깊은 에너지 골짜기(local minima)에 갇혀 무의미한 연산 시간을 낭비하는 치명적인 단점이 존재함.
- 생성 AI를 위한 문제 정의 (Problem Formulation)
- Task: 값비싼 MD 시뮬레이션을 대체하여, 극히 적은 비용으로 대리 분포(surrogate distributions)를 생성하는 모델 구축.
- Training: 일반화(generalization)를 위해 다양한 단백질 시퀀스의 MD 궤적(trajectories) 데이터를 활용하여 단일 생성 모델을 학습.
- Inference (Zero-shot): 학습에 사용되지 않은 완전히 새로운(unseen) 단백질의 시퀀스와 초기 구조 $x(0)$가 주어졌을 때, 실제 MD 시뮬레이션 결과와 통계적으로 구분할 수 없는 구조적 앙상블을 생성해 내는 것(물리적 관측값 $\phi$ 기준).
쉬운 설명 :
이 섹션은 **"우리가 AI로 정확히 어떤 문제를 풀려고 하는가?"**를 정의하는 바탕 설정 단계입니다.
단백질이 어떻게 움직이는지 알아내는 가장 정석적인 방법은 물리 법칙을 하나하나 계산하는 MD(분자동역학) 시뮬레이션입니다. 하지만 이 방식은 엄청난 문제가 있습니다. 단백질이 안정된 상태(에너지 골짜기)에 한 번 빠지면, 다른 상태로 넘어가기 위한 높은 산(에너지 장벽)을 넘는 데 너무 오랜 시간이 걸립니다. 비유하자면, 1초 단위로 걸음걸이를 계산해서 서울에서 부산까지 가야 하는데, 대전쯤에서 길을 잃고 빙빙 도느라 슈퍼컴퓨터를 몇 달씩 돌려도 결과를 얻기 힘든 셈입니다.
그래서 연구진은 생성 AI를 도입하기로 합니다. 이 AI의 임무는 다음과 같습니다.
여러 단백질의 기존 움직임 데이터를 보고 학습한 뒤, **"처음 보는 새로운 단백질"**을 던져주었을 때 1초 단위로 무식하게 계산하는 대신 통계적으로 단백질이 어떤 형태들을 취할지 순식간에(대리 분포 샘플링) 그려내는 것입니다.
결과적으로 AI가 만들어낸 단백질의 다양한 형태(앙상블) 모음이, 슈퍼컴퓨터를 수개월 돌려서 얻은 결과물과 통계적으로 구별할 수 없을 정도로 똑같게 만드는 것이 최종 목표라고 선언하고 있습니다.
3 MD Emulators and the challenges of fixed lag time transitions
MD sampling을 설명하는 핵심 객체는 lag time $\tau$와 연관된 transition density $y \sim p_\tau(y|x)$이며, 이는 시간 $\tau$ 내에 state $x$가 state $y$로 진화할 확률을 나타냅니다. 구체적으로, 이는 input frame $x(t)$가 주어졌을 때 미래의 transitions $x(t+\tau)$를 조사하여 MD trajectories로부터 추정됩니다. MD Emulators (MD-Emus)는 MD 데이터로부터 $p_\tau(\cdot|x(t))$를 근사하도록 trained된 conditional generative models의 한 클래스입니다. 구체적으로, MD-Emus는 $x(t)$가 주어졌을 때 미래의 conformations $y \sim p^\theta_\tau(y|x(t))$를 generate하도록 learn합니다. 일반적인 training 접근법에는 Normalizing Flows, Flow Matching, 또는 Score Matching이 포함됩니다.
이러한 models는 sequence와 input conformation $x(t)$를 조건으로 하는 neural network $v_\theta$를 train하여, MD 데이터에서 관찰된 transitions의 empirical distribution과 일치시킵니다. Jing 등은 MDGen에서 한 번에 다음 $K$개의 미래 conformations를 generate함으로써 이러한 접근 방식을 개선했습니다. 명시적으로, MDGen은 $y_0 = x$를 조건으로 하여 joint density $y = (y_1, \dots, y_K) \sim p_{\tau,K}(y|x) = \prod_i p_\tau(y_{i+1}|y_i)$에서 sample하는 방법을 learn합니다. 다음 내용에서는 $p_{\tau,K}$를 근사하도록 trained된 MD-Emu가 주어졌다고 가정합니다.
3.1 Limitations of MD-Emus
Training.
MD trajectories로부터 learning할 때의 주요 과제 중 하나는 에너지 최소값(minima) 내에서의 빈번하지만 정보가 없는(uninformative) transitions와 서로 다른 최소값 사이의 드물지만 핵심적인 transitions 간의 데이터 불균형입니다. 상황을 더 구체적으로 설명하기 위해, 단백질 MD trajectory가 높은 에너지 장벽(high-energy barriers)으로 분리된 folded 및 unfolded conformations와 같은 두 개(또는 그 이상)의 macroscopic states $S_A$ 및 $S_B$를 방문하는 경우를 고려해 보겠습니다. $S_A \to S_B$ transitions는 conformational 유연성과 기능적 dynamics를 보여주기 때문에 드물지만 매우 중요합니다. 그러나 길이가 $K\tau$인 주어진 trajectory 구간에서 시스템은 에너지 장벽을 넘기보다는 단일 state에 머무를 가능성이 훨씬 높습니다. 결과적으로, MD-Emus를 위한 training batches는 일반적으로 고주파수(high-frequency), 저정보(low-information)의 intra-state transitions에 의해 지배됩니다.
이러한 데이터 불균형은 의미 있는 state changes를 설명하는 conditional distributions를 learn하는 model의 능력을 제한합니다. 예를 들어, folded에서 unfolded states로 transition하는 방법을 learn하려면 model은 데이터에서 실제 transitions $S_A \ni x(t) \to x(t + K\tau) \in S_B$를 관찰해야 하지만 이러한 이벤트는 드뭅니다. 이러한 희소성은 training 데이터의 다양성과 효율성을 제약하고 generalization에 영향을 미치는데, 이는 MD-Emus가 large conformational changes보다는 fine-grained temporal resolution을 복제하도록 learn하기 때문입니다.
Inference.
긴 trajectories를 generate하려면 MD-Emus를 autoregressively하게 사용해야 합니다. 이는 error accumulation으로 이어져, samples가 data manifold에서 점진적으로 멀어지게 만들 수 있습니다. Nam 등은 이를 해결하기 위해 refinement step을 도입했지만, 이는 두 번째 network를 train해야 하는 비용이 수반됩니다. 게다가, 더 큰 $K$를 사용하는 것이 문제를 완전히 해결하지는 못하는데, 필요한 samples의 수가 더 큰 단백질에 대해 training 중에 가능한 최대 창(window)을 초과하는 경우가 많기 때문입니다.
Conclusion.
MD-Emus가 직면한 과제는 fixed lag time transitions가 종종 uninformative하며 서로 다른 에너지 최소값에 걸친 의미 있는 conformational changes를 capture하지 못한다는 점에 기인합니다. 이러한 limitations를 극복하기 위해, 우리는 먼저 고주파수 정보를 버리기 위해 MD 데이터에 coarse-grained representation을 적용해야 합니다. 이 목표를 위해 우리는 Markov State Models (MSMs)에 의존하고 frames가 아닌 states 사이의 transitions를 직접 learn하기 위해 generative flows의 새로운 클래스를 소개합니다. 핵심적인 이점은 trajectories가 metastable states로 clustered되고 나면, 더 이상 고정된 해상도에서 time series를 슬라이싱하여 얻은 $x_t \to x_{t+\tau}$ 형태의 frame pairs에 대해서만 training하도록 제한되지 않는다는 것입니다. 대신, 연속적인 MSM states에 할당된 어떠한 frame pairs도 training에 사용될 수 있어, informative transitions의 수와 다양성을 크게 증가시킵니다.
논문 3 MD Emulators and the challenges of fixed lag time transitions 요약 정리노트 (AI 연구자용)
- MD-Emus의 기본 접근법
- 모델은 고정된 시간 간격($\tau$)에서 미래의 프레임을 예측하는 transition density $p_\tau(y|x)$를 근사하도록 학습됨 (Flow Matching, Score Matching 등 활용).
- 기존의 SOTA 방식(MDGen 등)은 한 번에 $K$개의 미래 프레임을 함께 예측하는 joint density $p_{\tau,K}$를 학습하여 성능을 높이고자 했으나, 고정된 시간 간격($\tau$) 자체에서 오는 근본적 한계는 극복하지 못함.
- 한계점 1: Training - 극심한 데이터 불균형 (Data Imbalance)
- 원인: MD trajectories의 대부분은 하나의 에너지 최소값(minima) 내에서 발생하는 고주파수의 무의미한 미세 진동(intra-state transitions)으로 채워져 있음. 반면, 높은 에너지 장벽을 넘어야 하는 폴딩/언폴딩 같은 핵심적인 거시적 상태 변화(inter-state transitions)는 극히 드묾.
- 결과: 모델의 training batches가 정보가가 없는 고주파 데이터로 도배됨. 따라서 모델은 거대한 구조적 변화(large conformational changes)를 학습하기보다는, 단순히 짧은 시간 해상도(fine-grained temporal resolution)에서의 미세한 움직임만 복제하도록 과적합되는 경향이 발생하여 일반화 능력이 떨어짐.
- 한계점 2: Inference - 자기회귀적 오차 누적 (Error Accumulation)
- 원인: 긴 궤적(long trajectories)을 생성하기 위해서는 모델을 autoregressively하게 반복 호출해야 함.
- 결과: 반복 호출 과정에서 오차가 누적되어, 샘플들이 원래의 data manifold에서 점진적으로 벗어나는 표류(drift) 현상이 발생함. $K$값을 키우거나 별도의 보정(refinement) 네트워크를 추가하는 우회책은 메모리 한계나 추가적인 학습 비용 문제로 인해 근본적인 해결책이 되지 못함.
- 패러다임의 전환: 상태(State) 기반 학습으로의 도약
- 해결책 제시: 고주파 노이즈를 버리기 위해 MD 데이터에 Markov State Models (MSMs)를 적용하여 coarse-grained representation으로 압축.
- 효과: 프레임 간의 고정된 시간($\tau$) 전이가 아닌, metastable states 간의 transitions를 직접 학습하는 generative flows를 도입. 이제 연속된 상태로 분류된 '어떤 프레임 쌍'이든 학습에 사용할 수 있게 되어, 유의미한(informative) 전이 데이터의 양과 다양성을 폭발적으로 증가시킴.
쉬운 설명 :
이 섹션은 **"기존 AI(MD-Emus)는 왜 실패할 수밖에 없었는가?"**에 대한 뼈아픈 분석입니다.
비유하자면, 사람의 하루 일과를 관찰하여 시뮬레이션하는 AI를 만든다고 가정해 보겠습니다.
기존 방식(MD-Emus)은 **'무조건 1초 간격으로 연속 촬영한 사진'**으로만 AI를 가르쳤습니다. 그런데 사람이 하루 중 가장 많은 시간을 보내는 것은 '침대에서 자는 시간'입니다. 결국 AI가 받아보는 사진의 99%는 '침대에서 뒤척이는 모습'뿐이고, 정작 중요한 이벤트인 '침대에서 일어나 출근하는 모습'은 찰나의 순간이라 거의 주어지지 않습니다.
그 결과, 기존 AI는 **'침대 위에서 1초마다 어떻게 뒤척이는지(고주파 정보)'**는 기가 막히게 잘 흉내 내지만, **'침대에서 일어나 회사로 가는 큰 변화(구조적 변화)'**는 배우지 못합니다. 게다가 하루 전체를 시뮬레이션하려고 1초 단위 예측을 수만 번 반복하다 보니(자기회귀 추론), 나중에는 엉뚱한 곳으로 길을 잃어버리게 됩니다(오차 누적).
그래서 이 논문이 내린 결론은 **"1초 간격의 촘촘한 사진을 버리자"**는 것입니다. 대신 사람의 하루를 **[침대], [회사], [식당]이라는 큼직한 '상태(Markov State)'**로 묶어버립니다. 그리고 AI에게 1초 뒤의 모습을 예측하라고 시키는 대신, **"[침대] 상태에서 [회사] 상태로 어떻게 넘어가는가?"**라는 크고 의미 있는 변화만 집중적으로 가르치자고 방향을 제시하는 부분입니다.
4 The new class of MSM Emulators
Markov State Models (MSMs)는 높은 빈도의 시간 신호(high-frequency time signal)를 제거하면서도 긴 시간 규모의 통계(long-timescale statistics)를 포착하는 coarse-grained representation을 제공합니다. MSMs에서 프레임 $x(t)$는 이산적인 상태(discrete states) $S_1, \ldots, S_M$에 할당되며, 역학(dynamics)은 이 상태들에 대한 Markov chain으로 모델링됩니다. 시간 간격 $\tau$가 주어지면 전이 행렬(transition matrix) $T$를 추정하는데, 여기서 $T_{ij}$는 시간 $\tau$ 내에 $S_i$에서 $S_j$로 전이될 확률입니다.
명시적으로, 이는 다음을 만족하는 확률 밀도(probability density) $p_T(\cdot|x(t))$를 고려함을 의미합니다.
가장 단순한 형태에서 MSMs는 각 상태 내에서 균일한 밀도(uniform density)를 가정합니다. 즉, $x(t) \in S_i$와 $y \in S_j$가 주어졌을 때, $p_T(y|x(t))$는 클러스터의 동일성(identity)에만 의존하고 특정 conformations에는 의존하지 않습니다. 이러한 coarse-graining은 제한된 전이 데이터에서도 긴 시간 규모의 역학에 대한 강력한 추정을 가능하게 합니다. 원칙적으로 이산적인 상태 ${S_i}$에 걸쳐 짧은 MD 시뮬레이션(길이 $\tau$)을 초기화하고 $T$를 통해 그들의 역학을 전파할 수 있습니다. 그러나 기초가 되는(보이지 않는) MSM의 상태에서 샘플링하는 것은 여전히 과제로 남아있습니다. 우리는 이 문제를 해결하는 generative models 클래스를 제안합니다.
Definition 1 (MSM Emulators)
MSM Emulators (MSM-Emus)는 $p_T(\cdot|x(t))$에서 conformations를 샘플링하도록 훈련된 generative models입니다. 여기서 $T$는 주어진 MSM의 Markov chain 전이 행렬입니다.
프레임 $x(t) \in S_i$에 대해 MSM은 후속 상태 $j \mapsto T_{ij}$에 대한 범주형 분포(categorical distribution)를 정의합니다. 우리는 $p_T(\cdot | x(t))$를 그에 따른 MD 프레임에 대한 혼합 분포(mixture distribution)로 해석합니다. 먼저 $T_{ij}$에 따라 목적지 상태 $S_j$를 샘플링한 다음, $S_j$에 할당된 MD 프레임의 경험적 앙상블에서 conformation을 샘플링합니다. 따라서 MSM-Emus는 정확한 프레임 간 MD 전이 밀도(frame-to-frame MD transition density)를 학습하는 것이 아니라, 이 MSM에 의해 유도된 혼합 분포에서 conformations를 추출하도록 훈련됩니다. MSM-Emus는 MSM 상태에만 의존하는 분포 $p_T(\cdot|x(t))$를 매칭함으로써 MD-Emus에 영향을 미치는 데이터 희소성 및 불균형 문제의 대부분을 우회합니다.
- Increased sample diversity: MSM-Emus는 시뮬레이션에서 관찰된 특정 전이로부터 학습 목적(learning objective)을 분리합니다. 프레임 간 전이 $x(t) \to x(t + \tau)$를 학습하는 대신 상태 간 전이(state-to-state transitions) $S_i \to S_j$를 학습하여 더 넓은 일반화(generalization)를 가능하게 합니다. 고정된 지연 시간 $\tau$에서 정확한 시간적 역학을 복제하려는 MD-Emus와 달리, MSM-Emus는 MSM에 의해 식별된 거시적 전이(macroscopic transitions)를 포착하는 데 중점을 둡니다.
- Fast exploration: 추론 시 MSM-Emus는 세밀한 시간적 역학에 얽매이지 않고 샘플 $y \sim p_T(y|x(0))$를 생성하여 에너지 지형(energy landscape)을 탐색할 수 있습니다. 이는 샘플링 효율성을 크게 향상시킵니다. 또한 이제 많은 conformations를 병렬로 생성할 수 있으므로, MSM-Emus는 자기회귀 호출(autoregressive calls)로 인한 복합 오류(compounding errors)를 줄입니다.
MSM-Emus는 MSM에 의해 인코딩된 coarse-grained 역학을 보존하도록 설계되었지만, 고정 지연 시간(fixed-lag) MD-Emus와 동일한 수준의 정확도로 MD 궤적의 모든 세밀한 역학적 observables를 충실하게 재현하도록 의도된 것은 아닙니다.
4.1 A representative framework: Markov Space Flow Matching
우리는 MSM-Emu 클래스의 새롭고 대표적인 프레임워크인 Markov Space Flow Matching (MarS-FM)을 소개합니다.
Constructing MSMs
MarS-FM의 특정 버전은 먼저 MSM을 구축하여 인스턴스화됩니다. 이를 위해 표준 도구를 사용합니다. 상태 ${S_i}$는 집단 변수(collective variables)의 저차원 공간에서 정의됩니다. 차원 축소를 위한 일반적인 접근법은 Time-lagged Independent Component Analysis (TICA)입니다. 대안으로 회전 반경 및 이차 구조 비율과 같은 observables에 직접 클러스터링을 적용할 수 있습니다. 두 가지 핵심 관찰 사항을 강조할 가치가 있습니다.
첫째, MSM 구축은 오프라인 전처리 단계로 한 번 수행됩니다. 이러한 도메인별 상태 공간과 전이 행렬은 훈련 내내 고정되어 계산 오버헤드를 최소화합니다. 둘째, MarS-FM은 MD에서 직접 관찰된 전이를 넘어 MSM 상태 간을 보간(interpolate)하는 법을 학습하므로 데이터 가용성을 희생하지 않고 MD-Emus보다 더 큰 $\tau$를 선택할 여유가 있습니다. 훈련 데이터의 전처리로 MSM이 정의된다는 점을 강조합니다. 추론 중에는 MSM 정보가 제공되지 않습니다.
Representation
단백질 conformations를 생성하기 위해 MarS-FM은 동일한 SE(3) 표현을 채택합니다. 프레임 $x(t)$의 각 잔기(residue) $\ell$은 회전(rotation)을 설명하는 단위 쿼터니언(unit quaternion) $q^\ell(t)$와 이동(translation) $r^\ell(t)$, 그리고 $k$개의 비틀림 각도(torsion angles) $\theta^\ell_k(t)$로 표현됩니다. 입력 $x(t)$가 주어지면 목표(target) conformation $y$는 회전-병진 공간(roto-translational space)에서의 오프셋으로 표현됩니다. 소스 분포를 목표 분포 $p_T(\cdot|x(t))$로 이동시키는 벡터 필드 $v_\theta$를 모델링하기 위해, 우리는 MDGen 아키텍처를 기반으로 구축합니다.
Training objective
우리는 Flow Matching을 사용하여 MarS-FM을 훈련시키며, 이는 소스 분포 $p_0 = \mathcal{N}(0, 1)$을 목표 분포 $p_1(\cdot|x(t)) = p_T(\cdot|x(t))$로 이동시키는 시간 종속 벡터 필드 $v_\theta$를 학습합니다. 구체적으로 입력 프레임 $x(t) \in S_i$가 주어지면, 먼저 MSM 전이 확률 $j \mapsto T_{ij}$에 따라 목적지 상태 $S_j$를 추출한 다음, $S_j$에 할당된 MD 프레임 중에서 목표 MD 프레임을 선택합니다. 실제로는 $S_j$에 있는 유한한 집합의 MD 프레임에서 균일하게 샘플링합니다.
결정적으로 우리는 특정 프레임 $x(t) \in S_i$에 조건을 부여하기 전에 먼저 각 상태 $S_i$를 균일하게 선택합니다. 이러한 상태 기반(state-based) 샘플링은 훈련 중에 드문 상태(rare states)가 더 자주 나타나도록 보장합니다.
Sampling
추론 시에 보이지 않는 단백질(unseen protein)의 시퀀스 $a$와 입력 conformation $x(0)$가 있다고 가정하고, 두 가지 샘플링 전략을 탐색합니다.
- Tree Sampling: 계층적 샘플링 체계에 따라 MarS-FM을 적용합니다. 먼저 $n$개의 프레임을 병렬로 생성하고, 각 생성된 프레임에 대해 다시 $n$개의 프레임을 생성하여 트리의 깊이를 확장합니다.
- MarS-FM $\oplus$ MD-Emu: MarS-FM과 MDGen을 결합합니다. 먼저 MarS-FM을 사용하여 conformations를 샘플링한 다음, MDGen을 사용하여 이러한 각 지점에서 개별적으로 더 짧은 궤적을 생성합니다.
두 경우 모두 대부분의 conformations를 병렬로 생성할 수 있어 자기회귀 샘플링을 줄입니다. 결정적으로 MarS-FM의 샘플은 시간적 역학에서 분리되어 있기 때문에 목표 분포를 더 효율적으로 탐색할 수 있습니다.

Figure 2: MD 구조의 클러스터링 및 거시적 상태 전이
이 그래프는 단백질의 복잡하고 미세한 움직임(고주파 정보)을 어떻게 **'의미 있는 큼직한 상태(MSM states)'**로 묶어내는지(Coarse-graining)를 보여주는 실제 데이터 시각화입니다.
- 그래프의 축 (평가 기준):
- X축 (Radius of gyration): 단백질의 '회전 반경'입니다. 값이 작을수록 단백질이 둥글게 뭉쳐 있고(Folded), 클수록 길게 풀려 있음(Unfolded)을 의미합니다.
- Y축 (Secondary-structure fraction): 2차 구조(알파 나선, 베타 면 등)의 비율입니다. 값이 높을수록 단백질 고유의 뼈대 구조가 잘 잡혀 있고, 낮을수록 구조가 풀려 있음을 뜻합니다.
- 히트맵과 붉은 점 (Cluster Centres):
- 배경의 파란색/보라색 영역은 단백질이 자주, 안정적으로 머무는 상태(에너지 최소값)를 나타냅니다.
- 이 영역들의 중심에 찍힌 붉은 점들이 바로 논문에서 말하는 '이산적인 상태(Discrete States)' 즉, 클러스터의 중심입니다. 1프레임 단위의 움직임을 버리고, 이 붉은 점들을 징검다리 삼아 학습을 진행하게 됩니다.
- 화살표와 숫자 (Markov chain transitions):
- 가장 왼쪽 위(단백질이 똘똘 뭉쳐진 상태)의 한 붉은 점에서 다른 점들로 향하는 점선 화살표와 숫자가 있습니다.
- 이는 1초 뒤의 미세한 움직임을 예측하는 것이 아니라, **"현재 폴딩된 상태에서 다음 $\tau$ 시간 뒤에 어느 쪽(더 풀어진 상태 등)으로 이동할 확률이 얼마인가?"**를 보여주는 Markov 전이 확률(Transition probability)입니다.
- 그래프 위쪽에 겹쳐진 단백질 3D 구조 그림을 보면, 왼쪽은 구조가 잘 잡힌 형태(Folded)이고 오른쪽 끝으로 갈수록 선처럼 길게 풀어진 형태(Unfolded)임을 알 수 있는데, MSM이 이런 **'거대한 구조적 변화'**를 성공적으로 포착하고 있음을 증명합니다.

Figure 3: MarS-FM의 계층적 샘플링 (Tree Sampling)
이 그림은 MarS-FM 모델이 새로운 단백질 구조를 생성해 낼 때(추론 과정) 사용하는 효율적인 '트리 샘플링(Tree Sampling)' 방식을 묘사하고 있습니다.
- 출발점 (붉은색 X): 샘플링이 시작되는 초기 단백질 구조입니다.
- 1차 샘플링 (굵은 녹색 점선): * 기존 모델들처럼 선을 긋듯 1프레임씩 쫄래쫄래 따라가는 것이 아닙니다.
- 출발점에서 Markov 전이 확률에 따라 서로 다른 여러 개의 상태(구덩이)로 단번에 뛰어넘는 샘플들($y_1, \ldots, y_n$)을 '병렬로(동시에)' 생성합니다.
- 2차 샘플링 (얇은 녹색 점선):
- 1차로 도달한 각각의 새로운 지점들에서, 또다시 여러 갈래로 뻗어나가는 다음 상태의 샘플들을 병렬로 생성합니다. 마치 나뭇가지가 뻗어나가는(Tree) 형태입니다.
- 핵심 의미:
- 이 방식은 꼬리에 꼬리를 무는 자기회귀(Autoregressive) 방식을 대폭 줄여주기 때문에 오차가 눈덩이처럼 불어나는 현상(Error accumulation)을 방지합니다.
- 또한 여러 갈래로 동시에 탐색을 진행하므로, 깊은 구덩이에 갇히지 않고 단백질의 전체 에너지 지형(Energy landscape)을 **훨씬 빠르고 폭넓게 탐색(Fast exploration)**할 수 있음을 직관적으로 보여줍니다.
논문 4 The new class of MSM Emulators 요약 정리노트 (AI 연구자용)
- MSM-Emus의 핵심 패러다임 (Core Concept)
- 목표의 재정의: 기존 MD-Emus처럼 특정 프레임 간의 정확한 전이 밀도(frame-to-frame transition density)를 흉내 내는 것을 포기함.
- 새로운 타겟: Markov State Model (MSM)이 계산해 낸 상태 간 전이 확률(transition matrix $T$)을 기반으로, 결과적인 혼합 분포(mixture distribution) $p_T(\cdot|x(t))$에서 샘플링하도록 모델을 훈련함.
- 왜 이 방식이 근본적인 문제를 해결하는가? (Key Advantages)
- 폭발적인 Sample Diversity: 궤적(trajectory)의 시간 순서에 얽매이지 않음. 특정 상태 $S_i$에 속한 아무 프레임과 $S_j$에 속한 아무 프레임을 짝지어 학습 데이터로 쓸 수 있음. 이를 통해 폴딩/언폴딩 같은 극히 드문 변화도 훈련 데이터로 다량 확보 가능.
- Fast Exploration: 세밀한 시간 역학($\tau$)을 건너뛰고 거시적 상태 사이를 도약함. 수백 개의 샘플을 병렬로 생성할 수 있어, 자기회귀(autoregressive) 추론 시 발생하는 치명적인 오차 누적(compounding errors)을 차단함.
- MarS-FM 프레임워크 핵심 디테일 (Implementation of MarS-FM)
- Offline Pre-processing: 차원 축소(TICA)나 구조적 지표를 활용해 MSM을 미리 구축함. 이는 훈련을 위한 '가이드'일 뿐, 추론(Inference) 단계에서는 MSM 정보가 전혀 필요 없음 (Zero-shot 가능).
- State-based Sampling (가장 중요한 훈련 기법): Flow Matching의 목표 타겟을 설정할 때, 궤적에서 프레임을 무작위로 뽑는 것이 아님.
- 상태 $S_i$를 먼저 균일하게(uniformly) 샘플링함.
- 그 상태 내에서 프레임 $x(t)$를 선택하고, $T$ 행렬에 따라 목적지 상태 $S_j$를 결정함.
- 이 과정을 통해 빈도가 압도적으로 높은 '무의미한 상태'와 극히 드문 '희귀 상태(rare states)'가 훈련 중에 동등한 비율로 노출되도록 강제함. 기존 MD 데이터의 극심한 불균형(Data Imbalance)을 완전히 박살 내는 핵심 트릭.
- 추론 및 샘플링 전략 (Inference Strategies)
- Tree Sampling: 한 점에서 여러 상태의 프레임을 병렬로 생성하고, 그 생성된 프레임들에서 다시 여러 프레임을 병렬로 생성하는 계층적 방식. 탐색 속도와 범위를 극대화함.
- MarS-FM $\oplus$ MD-Emu: 거시적 구조 변화는 MarS-FM으로 크게 도약(jump)시키고, 도달한 각 지점에서의 미세한 시간적 역학(local dynamics)은 기존 MDGen을 붙여서 시뮬레이션하는 완벽한 하이브리드 전략 제안.
쉬운 설명 :
이 논문이 제안하는 MarS-FM의 학습 방식을 비유하자면 **"AI에게 1cm씩 걷는 법을 가르치는 대신, 공간이동 포탈을 여는 법을 가르친 것"**과 같습니다.
기존 AI들은 단백질 시뮬레이션 비디오를 처음부터 끝까지 틀어놓고 1초마다 어떻게 변하는지를 학습했습니다. 그런데 단백질은 하루 종일 가만히(안정된 상태) 있다가 0.001초 만에 확 접혀버립니다. 비디오를 그대로 보고 배운 AI는 가만히 있는 모습만 주구장창 학습하게 되니, 나중에 새로운 단백질을 줘도 "그냥 가만히 있겠지 뭐"하고 커다란 변화를 예측하지 못합니다.
그래서 연구진은 아예 비디오를 해체해 버렸습니다.
- 단백질의 모습들을 비슷한 것끼리 묶어서 **'여러 개의 바구니(상태)'**에 담습니다.
- AI를 학습시킬 때, 비디오를 틀어주는 대신 각 바구니에서 골고루 샘플을 하나씩 꺼내서 보여줍니다. (이것이 State-based sampling입니다.)
- "A 바구니에 있던 단백질이 B 바구니로 변할 확률은 10%야. 자, 이제 A에서 B로 변하는 모습을 그려봐!" 하고 시킵니다.
이렇게 바구니 단위로 골고루 학습시키니, 어쩌다 한 번 일어나는 희귀한 '접힘/풀림' 현상도 AI는 질리도록 연습하게 됩니다.
그 결과, 새로운 단백질을 만나면 이 AI는 1초 뒤, 2초 뒤를 찔끔찔끔 예측하지 않습니다. 단번에 커다란 구조적 변화를 예상하여 나뭇가지가 뻗어나가듯(Tree Sampling) 여러 형태의 결과물을 펑펑 쏟아냅니다. 미세한 떨림까지 확인하고 싶다면? 크게 이동시킨 다음 그 자리에서 기존 AI(MD-Emus)를 켜서 꼼꼼히 살펴보면 됩니다. 이것이 이 논문이 제안하는 혁신적인 패러다임입니다.
별점 3점 / 5점
“백본은 사실상 MDGen 계열 그대로고, 진짜 기여는 MSM 기반 학습쌍 구성과 objective 재정의에 있다. 방법론적 새로움은 약하지만, 단순 데이터 갈아끼우기 이상은 해서 3점.”